ALS: Perspectives and Experiences
ALS: Perspectives and Experiences
Dr. Hassan Jazayeri*
*Correspondence to: Dr. Hassan Jazayeri, Neurologist, Albania.
Copyright
© 2024 Dr. Hassan Jazayeri, This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 20 June 2024
Published: 01 July 2024
ALS: Perspectives and Experiences
Introduction
Recently, on one of the sites, I saw a short dance video of a mother who, with the help of her son, tried to dance in a crowd of 150 people. This mother, who had advanced ALS, symbolically danced with the help of her son and her husband. She achieved her long-time dream of dancing at her son's wedding. The eyes of those present in this scene were full of tears, tears of joy that the patient with advanced ALS has achieved her wish. Ten days later, Terry's mother died. But this memory remained between all of them and those who later saw her film, including me.
Figure 1
After watching this short film, I also remembered my patients, who had the same disease (ALS), and had already died.
Considering that ALS disease is very rare and affects 2.5% of every 100,000 people, I considered it my duty to publish their reports to colleagues, students and medical researchers, so that they can share their experiences with these patients. To be used.
During my forty years of neurology work, I have encountered ALS patients in many countries, especially in the Middle East, and I have closely followed the work of about 11 of these patients so far. I remember well the death of three of these patients.
ALS was first described in 1869 by the French neurologist Jean-Martin Charcot and hence is also known as Charcot disease. However, after the announcement of this disease in the United States of America, it is also known as Lou Gehrig's disease. Lou Gehrig's was a well-known baseball player who was diagnosed with this disease in 1939, and this disease was named after Logherik. Actually, amyotrophic lateral sclerosis is a very aggressive and debilitating type of motor neuron disease (MND).
Unlike multiple sclerosis patients, who live an almost normal life, ALS patients die quickly in a short period of time. The cases that I will reread in this report died within six months of the diagnosis and confirmation of their disease.
But I must admit that Mr. Steve Hawking was an exception among ALS patients that I don't imagine will be repeated again in the history of medicine.
And but Stephen Hawking:
He was one of the ALS sufferers who, despite the severe deterioration and progress of the disease, was able to become one of the world's leading physicists and cosmologists by relying on his abundant intelligence and talent.
Figure 2
He was the research director of the Center for Theoretical Cosmology at the University of Cambridge, who, for more than forty years, wrote books such as: History of Time: From the Big Bang to Black Holes, The Big Plan and participated in major scientific conferences as a prominent figure in the field of physics and cosmos. Science was known. Hawking did a lot of research in the field of space black holes and was able to win the prize for pioneers of science in fundamental physics.
He, who was born on the 8th of January 1942 in the university city of Oxford, during the Second World War, after years of fighting with ALS, finally passed away in the early hours of Wednesday (March 14) at the age of 76. The world collapsed.
It is difficult to determine why some people develop the disease while others do not. However, researchers have some possible ideas.
ALS targets the motor neurons of the patient and in the process it disrupts the movement of voluntary muscles and the subsequent steps of smooth muscle function. These are the nerve cells that control important muscle activities, including4 breathing, speaking, swallowing, and walking. Over time, the loss of muscle control worsens. There is no cure for ALS, although research is ongoing. There are no preventive measures.
Pathogenesis
It is important to distinguish between the pathogenesis and pathophysiology of ALS. Because the mechanisms underlying each of these steps are likely to be different. This means that interfering with these mechanisms will likely require different approaches. Interventions to prevent the onset of disease are likely not the same as those needed to slow or stop its progression once it has begun. Prevention of ALS requires modification or elimination of factors that are part of the pathogenesis of the disease. A prerequisite is to identify potential risk factors for ALS.
Unlike the approach to understanding the pathophysiology of ALS, which relies on direct biological experiments, the mechanisms that lead to the onset of the disease (ie, pathogenesis) are inferred from strong circumstantial evidence. In patients with familial ALS, it is reasonable to assume that an abnormal gene or gene product is involved in the onset of the disease and may contribute to the spread of the disease, but having an abnormal gene is neither necessary nor sufficient to cause ALS.
Other factors must be hypothesized to intervene between birth and disease onset even in patients with familial ALS, as the disease does not appear to begin at birth and within a given family, the age of onset varies widely. Having normal copies of these genes does not prevent sporadic ALS.
The onset of ALS is a multistep process. Most likely, it occurs in a cortical motor neuron with or without its immediate environment, similar to the initiation of malignancies in dividing cells. The number of stages in sporadic ALS is 6, and less in carriers of the disease. Different primary genes require a different number of additional steps to initiate the disease. . The need for additional steps in patients who carry the gene explains why they are born with ALS are not affected and why not all carriers are affected ("incomplete penetrance").
Family history of this disease is found in about 5% of patients, and twin studies show a genetic contribution with a heritability of about 61%. Recently, up to 20% of patients presenting with sporadic ALS carry pathogenic or potentially pathogenic genes.
Prevalence:
The illness is relentlessly progressive, leading to death from respiratory paralysis; the median survival is from 3 to 5 years. There are very rare reports of stabilization or even regression of ALS. In most societies, there is an incidence of 1–3 per 100,000 and a prevalence of 3–5 per 100,000. It is striking that at least 1 in 1000 deaths in North America and Western Europe (and probably elsewhere) are due to ALS; this finding predicts that more than 300,000 individuals now alive in the United States will die of ALS. Several endemic foci of higher prevalence exist in the western Pacific (e.g., in specific regions of Guam or Papua New Guinea). In the United States and Europe, men are somewhat more frequently affected than women. Epidemiologic studies have incriminated risk factors for this disease including :
Gender: The ratio of ALS patients to these patients is 3:2 in terms of male to female, and the percentage of people affected are male. The cases I have had were all male patients, and I have never seen a female patient with ALS, neither in clinic visits nor in outpatient departments.
Race: 93% of people with ALS are white. All the patients under my supervision were all white and from the Middle East.
Aging: Although this disease can occur at any age, symptoms usually develop between the ages of 55 and 75. It can be said that it is very rare to develop it before the age of 30. The patients I personally monitored were in the age group of 55 to 65.
Family history: A small percentage of ALS cases are passed down from the family. But in my patients, we could not fundamentally address this issue because in many cases their families were not available to take histories or medical examinations. In addition, the patients themselves did not express any information about this type of disease in their relatives.
Risk factors
What factors should be considered as risk factors for ALS disease, there are still different opinions among experts. But in general, most scientists and neurologists agree on the following.
Smoking: It is believed that smoking is the only possible factor that may increase the incidence of ALS. All three patients I had, who died with the same disease, had a history of smoking for a long time. However, one of them had completely quit smoking twenty years before he died.
Military service: Studies have shown that military veterans, especially those who served during the 1991 Persian Gulf War, have a higher chance of developing ALS. The cause is still unclear, but may include exposure to chemicals or metals, injuries, infections, or vigorous physical activity. Those who were in the Persian Gulf War are more likely to develop ALS than other veterans. All three of the patients I had, although they themselves had not directly participated in the Persian Gulf War in Iraq and had spent a long time in war-contaminated areas, were present.
Vigorous activity: The most famous person with ALS was baseball player Lou Gehrig, who died from it. Studies have shown higher odds among athletes who are very active. But these studies are not complete, so it is too early to say that being an athlete means that they have a higher chance of getting this disease.
Type of Job activities:
Several lines of work, including sports, cockpit, construction, farm, hairdressing, laboratory, veterinary, and welding, among many other occupations, have been reported to be more likely to develop ALS. These jobs often involve some form of contact with pesticides, metals, and chemicals. But common and essential risk has not been found.
Exposure to lead toxins and other chemicals may be associated with ALS, but no single factor has been consistently identified as the cause.
Other risk factors
Trauma, physical activity, living in rural areas, and alcohol consumption are not likely risk factors for ALS.
Clinical Finding
In 75-80% of patients, symptoms begin with organ involvement. Primary complaints in patients with lower limb onset are often as follows:
Movement signs:
Falling, stumbling, or clumsiness while running, loss of volume, strength or endurance of lower limb muscles, leg drop or foot drop.
Primary complaints starting in the upper limbs include the following:
Decreasing finger dexterity, cramping, stiffness, and weakness or atrophy of the inner muscles of the hand.
Wrist drop interferes with work performance
Autonomic symptoms in the medulla:
With the onset of medullary lesions that may develop in (20-25% of patients), the initial complaints of patients may be reported as follows:
Slurred speech, hoarseness, or decreased speech volume
Respiratory disorders:
One of the most important problems of patients is lung problems, and in patients, one of the causes of their death may be aspiration or suffocation during meals or breathing problems. In all three cases, the cause of their death have been reported as shortness of breath disorders and respiratory arrest.
Psychological Disturbances:
Psychological problems are one of the serious problems of these patients. Of course, in patients who have these problems in a non-progressive way, and in the process of treatment, they realize that radical treatment is not intended to solve their problem, the emergence of psychological problems is very effective. By the way, patients who accept this fact and adapt themselves, can do better. Use the opportunities obtained. While if they think about this issue more, their life span will automatically be shortened. Psychological symptoms in some ALS patients are as follows:
Involuntary laughing or crying, depression, impaired executive function and other cognitive changes, behavioral changes
Memory disorders:
Some patients show cognitive and behavioral changes due to prefrontal cortex involvement. In its severe form, these changes meet the criteria for Behavior Variant Fronto-Temporal Dementia. It occurs in 16% of patients with ALS. 33% will have behavioral or cognitive changes.
Characteristics of disease progression;
Manifestation of muscle atrophy, spasticity that may compromise walking and manual dexterity, muscle cramps, along with painful joint spasms caused by immobility.
Progression of myelopathy leads to the following:
Voice changes, stuffy nose and creating a weak and muffled sound quality. Finally, speech may be lost, swallowing problems, usually starting with liquids, drooling.
ALS is fatal to everyone who develops it, but the timeline is difficult to predict.
Clinical symptoms of my patients
Positive symptoms:
• These patients suffered from walking problems, so at first we could not distinguish whether they have discopathy problems or they might be MND. All three cases were completely rejected by performing spine MRI.
• Atrophy of the muscles of the lower limbs unilaterally and in a short period bilaterally, especially in the lower limbs with sensory disorders. In one of the patients, foot drop was very obvious without being able to find a reason for this problem.
• In all three of my patients, respiratory symptoms, especially shortness of breath, were very evident. So that at first it seemed that the patients had lung problems. But the examinations related to chest MRA and spirometry determined that no significant lesions were seen in the lungs. When the ALS diagnosis was confirmed, I was convinced that the patients' lung problems were caused by ALS
• I remember very well in all three cases that they died with respiratory disorders and under an oxygen tent. This was the time when the diagnoses were confirmed and sometimes respiratory disorders appeared, and the disease was considered to be progressing.
• In only one case, we encountered swallowing disorders, which were accompanied by speech problems, of course, to a small extent, which manifested itself in the form of slurring and slurred words.
• One of the problems they faced was the problem of appetite. All three of them had anorexia. Thus, in the last days of their lives, each of them had lost more than ten kilos, laboratory examination did not show that their anorexia was caused by a physical problem.
• All three of our patients were forced to use wheelchairs and wheelchairs for their movements almost two months before they died because they could not resolve their individual issues.
Negative symptoms:
These patients basically did not have these problems
• Psychological problems. It was very interesting to me that these patients did not face any psychological problems until the last moments of their lives, neither depression, nor stress, nor any psychological abnormalities. I believe that one of the reasons why these patients never faced psychological problems was the human relationships that friends and their relatives arranged with these patients. In many hours, they came to see these patients and helped them with their illness.
• Also, they did not have any sleep disorders and every now and then when they faced this problem, they solved the problem sporadically with lorazepam tablets of half a milligram.
• They did not know about their family, and in fact, none of them had a history of a disease similar to theirs, so that we could confirm the connection of their disease to the family.
• We were not able to perform genetic tests in any of these patients, so these things were not done at all
• The only possible diagnostic evaluations were MRI and EMG, of course, we did not find a valid lesion in any of these cases, and the EMG tests were more or less positive in both sensation and movement. Most of our sure information was the clinical symptoms of the patients, which clearly showed themselves in movement.
• We must emphasize that all three patients were unable to move even on the bed almost in the last days of their lives, and at the same time, respiratory problems were prioritized over other patients' complaints and problems.
• We did not face any significant ophthalmological problems. Despite the fact that the patients had no problems, we were able to perform OCT tests and clinical fundus examinations with ophthalmology specialists. Because in the first days, one of our differential diagnoses was multiple sclerosis. Although the age of onset of their disease It was also in contradiction with MS, but we had to do this to ensure a definite diagnosis
Physical Exams on A.L.S case:
_ UMN signs: Hyperreflexia, spasticity, Babinski’s sign, emotional lability (pseudobulbar affect)
_ LMN signs: Weakness, hyporeflexia, atrophy, diffuse fasciculation’s, flaccidity, cramps (calves)
_ Bulbar (ALS onset 20–30%): Tongue atrophy or fasciculation’s, dysarthria (flaccid or spastic), jaw jerk/gag (reduced or hyperactive), dysphagia
_ Respiratory: Exertional dyspnea, orthopnea
– Chronic ventilatory insufficiency: Heralded by fitful sleep, orthopnea, nightmares, morning headache, drowsiness, change in snoring
_ Cognitive: Frontotemporal dysfunction
_ El Escorial Criteria [C] (World Federation of Neurology): Progressive weakness or
electromyographic (EMG) signs in bulbar, cervical, thoracic, lumbosacral “regions”
– Definite ALS: UMN and LMN signs in 3 regions (bulbar + 2 spinal or 3 spinal)
– Probable ALS: UMN and LMN signs in 2 regions, some UMN rostral to LMN
Diagnosis
Diagnostic criteria for amyotrophic lateral sclerosis have been established by the World Federation of Neurology. Criteria vary depending on the level of certainty of the diagnosis, as shown in Table 9-11. Definitive diagnosis requires the presence of upper and lower motor neuron signs in the bulbar region and at least two other spinal regions (cervical, thoracic, or lumbosacral), or in three spinal regions. Alternative causes of signs and symptoms must be excluded.
A definitive diagnosis may not be possible with early-onset ALS. Confirmation of the disease may require a period of observation to document its progressive nature and rule out alternative diagnoses. MRI of the brain and spine, as well as EMG, are usually necessary for diagnosis. Neurologists may use other diagnostic methods such as EEG, or, genetic tests and lumbar puncture may be used
Figure 3
Complications:
Progressive inability to perform activities of daily living (ADLs), including the ability to eat without assistance, aspiration pneumonia, respiratory failure, complications from wheelchair use or hospitalization, including skin ulcers and Skin vents, deep vein thrombosis and pulmonary embolism.
Emotional and mental health care
There is no right way to react to ALS, and it's normal to go through a range of emotions after a diagnosis. You may feel angry, sad, or scared. Many people feel anticipatory grief—a strong sense of sadness about future losses.
Strong emotions such as shock may diminish over time, while other emotions, including death anxiety, may become stronger. These mood symptoms can affect you just as much as physical problems. The good news is that there are ways to increase your well-being, no matter what your circumstances.
Most people with ALS do not develop anxiety disorders or depression.
Treatment
Life extension is possible with Riluzole drug as well as ventilation and nutrition therapy. As a result of ALS, various stressful symptoms may develop that can be significantly reduced or completely controlled with appropriate medication. Physical therapy, occupational therapy, and speech therapy can also reduce symptoms and strengthen preserved motor functions. Individual and modern aids can reduce mobility and communication limitations and improve participation in private and social life. In ALS, breathing may become impaired due to weakness of the respiratory muscles. In this situation, mask ventilation is available. It is a mechanical respiratory aid that takes over part of the work of breathing that would otherwise be done entirely by the respiratory muscles. Mask ventilation is adjusted during a short hospital stay and can then be used independently at home. If the respiratory muscles are severely weakened, the work of breathing or coughing cannot be fully compensated by mask ventilation. Then, under certain conditions, respiratory weakness can be compensated through tracheostomy and mechanical ventilation (invasive ventilation therapy). Even with this "artificial ventilation", ALS progresses and the body can become completely paralyzed.
I prefer to conclude the treatment of this disease by using the latest valuable experiences of the Amyotrophic Lateral Sclerosis Society, who have published very positive experiences in this field, which, when we reviewed them, were very remarkable.
FDA-Approved Drugs for Treating ALS
There are currently six drugs approved by the U.S. Food and Drug Administration (FDA) to treat ALS and its symptoms:?Qalsody, Radicava, Rilutek, Tiglutik, Exservan and Nuedexta. Please consult your doctor or health care professional about which ones may be right for you.
In addition, research is being conducted all over the world to develop more treatments and a cure for ALS. Learn more about clinical trials.
Qalsody (tofersen)
Qalsody, also known as tofersen or BIIB067, was developed to treat ALS associated with a mutation in the superoxide dismutase 1 (SOD1) gene. The FDA approved Qalsody for use to treat SOD1-ALS in 2023. Learn more.
Radicava™ (edaravone)
The FDA approved Radicava™ in 2017, making it the first new treatment specifically for ALS in 22 years. An oral formulation was approved in 2022. Learn more.
Rilutek (riluzole, now generic)
This was the first FDA-approved drug available to treat ALS — in 1995. It inhibits glutamate release and prolongs life by approximately three months. Riluzole is the generic name of Rilutek.
Tiglutik (thickened riluzole)
The first and only thickened liquid form of?riluzole,?Tiglutik?was approved by the FDA in September 2018. This formulation contrasts with the oral pill form of?riluzole?that has been on the market for ALS for more than 20 years. It is designed to avoid potential problems of crushing tablets.??Learn more.
Exservan™ (riluzole oral film)
An oral film formulation of riluzole, Exservan was approved by the FDA in November 2019. This formulation of riluzole, which has been on the market for ALS for more than 20 years,?was developed for people with severe swallowing difficulties. The oral film is placed on top of the person’s tongue and dissolves, bypassing the need to swallow a pill or liquid.?
Nuedexta®
Approved by the FDA in 2011, Nuedexta® (dextromethorphan HBr and quinidine sulfate) is prescribed to help treat pseudobulbar affect (PBA), which is characterized by frequent, involuntary, and often sudden episodes of crying and/or laughing that are exaggerated and/or don’t match how the person truly feels. PBA occurs secondary to a variety of otherwise unrelated neurologic conditions.
RELYVRIO (AMX0035),
which is a combination of sodium phenylbutyrate and taurursodiol, was approved by the FDA to treat ALS in 2022. However, this medication was voluntarily removed from the U.S. and Canadian markets based on topline results from the phase 3 PHOENIX trial. As of April 4, 2024, RELYVRIO is no longer available to new patients. People living with ALS who were already receiving the therapy in the U.S. and Canada, where it was called Albrioza, were given the option to continue taking this medication after consulting with a health care professional.
UPDATE – APRIL 2024: Relyvrio was voluntarily withdrawn from the market by Amylyx following a phase 3 trial that failed to show it was effective. The ALS Association stands by its decision to push for early approval of Relyvrio given the promising phase 2 trial data and the safety of the treatment. At the time, we said that if it turns out to be ineffective, at worst, people living with ALS would have taken an ineffective therapy without risk of harm. If it was indeed effective, delaying access would have meant that people living with ALS would have lost two years of being able to take a life-extending therapy. In the interests of transparency and education, we are leaving this information up for future reference. People living with ALS need life-saving treatments and we are working as urgently as possible to advance the many more potential treatments in clinical trials.
Prognosis
There is currently no cure for ALS. However, many forms of treatment can increase life expectancy, reduce symptoms, and improve participation. Life with ALS is limited by two sets of symptoms: a severe swallowing disorder (and associated malnutrition) and decreased respiratory function (weak breathing or narrowing of the airways).
If swallowing is severely impaired, a feeding tube (percutaneous endoscopic gastrostomy, PEG) may be used to provide adequate nutrition (usually in addition to regular meals). With ventilation therapy, respiratory weakness or narrowing of the airways can be overcome and life can be prolonged.
Coping with amyotrophic lateral sclerosis (ALS) in everyday life
The use of assistive devices and "assistive technology" (such as communication systems, environmental controls, and robotic arms) is one of the most important measures that enable people with ALS to have a high level of private or social participation. Assistive devices are designed according to the personal needs of the patient and their relatives - according to the principle: as little as possible, but as much as necessary. There are significant individual differences in the treatment of ALS. One of the main factors that lead to the personal adjustment of the concept of treatment is the different attitudes of the affected people towards life-prolonging measures. Acceptance or rejection of life-enhancing measures does not create a "black and white picture" but is a different decision-making process that should be carefully documented. To facilitate decision-making and documentation, there are templates of living wills designed for common decision-making situations in ALS. These include decisions about nutritional care (eg, with a feeding tube), assistance with breathing (eg, with a respirator) and prolonging life through "artificial ventilation" (invasive ventilator therapy). If you have any questions about care, the German Brain Foundation and its specialists will be happy to help.
Conclusion
Although the prevalence of amyotrophic lateral sclerosis is very low. Once diagnosed, there is no treatment of choice to contain and control the progression of the disease. Most importantly, the speed of its progress is very significant. However, it is very necessary to perform multiple Para clinical methods to rule out many similar diseases and direct one of them towards ALS. These actions help us focus better on our subjects.
In working with these patients, I also encountered two interesting cases.
One of them is a 67-year-old man who has been diagnosed with ALS for more than 9 years, but it seems that his disease has stopped and is not progressing. His main problem is lack of balance and Weakness in the lower limbs is accompanied by muscle atrophy.
The second case is a 53-year-old man who only has hypotrophy in the thenar and hypothenar muscles of the right hand with the muscles of the forearm, which has reduced his muscle strength and efficiency, and this condition has remained stable and unchanged. I have not found a medical term to name for this term, and we ourselves use the established ALS in our medical group.
• For patients who are diagnosed with ALS, their vital threats, including respiratory distress, should be told and justified to all patients and co-workers.
• Active and passive physical therapies (depending on the patients' conditions) can be effective in reducing the patients' problems.
Reference
1. Clinical-Neurology-Aminoff-2018-10th-edition-extern.ir (Page 250-1)
2. Textbook of Clinical Neurology 1st Edition (Page 39)
3. 5-Minute_Neurology_Consult 2th edition (Page 64-65)
4. Harrisons-Principles-of-Internal-Medicine-2022-(Page 3410-3415)
5. Medically Reviewed by Christopher Melinosky, MD on April 14, 2023, Written by Kara Mayer Robinson, Keri Wiginton
6. Medically reviewed by Heidi Moawad, M.D. — Written by Verneda Lights and C.
Guthrie — Updated on December 18, 2023
7.https://www.als.org/navigating-als/living-with-als/fda-approved-drugs#:~:text=There%20are%20currently%20six%20drugs,may%20be%20right%20for%20you.

Figure 1

Figure 2
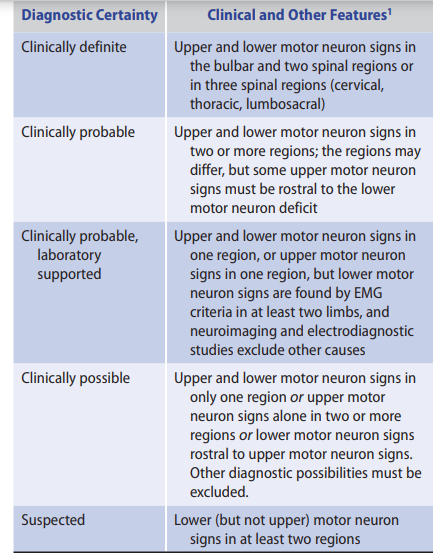
Figure 3