Embryonal Paratesticular Rhabdomyosarcoma in Child Diagnosed Accidentally After Trauma: A Case Report
Embryonal Paratesticular Rhabdomyosarcoma in Child Diagnosed Accidentally After Trauma: A Case Report
Raad Y Altahat *1, Mohammad F Hamidi *3, Hasan N Al-Haidari **1, Ibrahim K Alhmaisat 2,
Khalid Abu Jamose 4
1.Radiology department, Jordanian Royal Medical services, King Hussien Medical center, Amman, Jordan
2.Radiology department Alsaudi hospital, Amman, Jordan.
3.Urology department, Alsaudi hospital, Amman, Jordan.
4.Radiology department, Ministry of Health, Al-Basheer hospital, Amman, Jordan.
* These two authors contributed equally to this work
**Correspondence to: Hasan N. Al-Haidari, Radiology department, Jordanian Royal Medical services, King Hussien Medical center, Amman, Jordan.
Copyright
© 2024 Hasan N. Al-Haidari. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 14 August 2024
Published: 02 September 2024
Abstract
Paratesticular Rhabdomyosarcoma (RMS) is a rare cancerous tumour that develops from connective tissue in the spermatic cord and epididymis. It is widely regarded as the most common type of soft tissue sarcoma in children. Typically, it is diagnosed incidentally. This paper presents the case of an 8-year-old boy who experienced trauma to his groin and was subsequently diagnosed with an embryonal paratesticular rhabdomyosarcoma accidentally.
Embryonal Paratesticular Rhabdomyosarcoma in Child Diagnosed Accidentally After Trauma: A Case Report
Introduction
Paratesticular Rhabdomyosarcoma (RMS) is an uncommon and aggressive cancer that develops from mesenchymal tissue in the spermatic cord and epididymis. The embryonal subtype accounts for approximately 90% of paratesticular rhabdomyosarcomas [1,2,3]. The age of onset exhibits two distinct peaks the first peak occurring between the ages of 2 and 5 years, and the second peak during adolescence [5]. The clinical diagnosis of paratesticular rhabdomyosarcoma is often incidental, revealing an indolent intrascrotal mass [25,4]. Ultrasound is the primary imaging modality used to diagnose paratesticular rhabdomyosarcoma. CT, MRI and PET scan are used to detect distant metastasis. Its management necessitates a multidisciplinary approach. Localized forms have a better prognosis, and a multimodal treatment approach can result in high survival rates.
Case Report
8-year-old boy presented to the clinic with a history of trauma by his brother to genital area. The family noticed left hemiscrotal swelling after the previous trauma. When the patient was presented to the clinic, he had painless swelling in the left scrotal with no overlying skin changes, which was inseparable from the left testicle and non-reducible, extending to the inguinal area (Figure1 A).
He underwent scrotal ultrasound, which revealed well-capsulated, heterogeneous, highly vascular mass in the left testis with superior extension. The image suggested a left testicular tumor. Then tumor markers were sent, and the results were unremarkable.
Then, the decision to perform a scrotal MRI and the report showed a large mass seen in the left hemiscrotum extending to upper inguinal canal. There was a clear cleavage line observed from the left testis. It demonstrated low signal intensity on TIWI and high signal intensity on T2WI, showing marked diffusion restriction in ADC/DWI sequences, and heterogenous post-contrast enhancement. The mass causes mass effect and compresses the left testis, which showed normal signal intensity (Figure3 and 4). The MRI findings were suggestive of paratesticular tumor, with embryonal rhabdomyosarcoma being the top consideration in the differential diagnosis.
The patient underwent a left radical orchidectomy orchiectomy through an inguinal incision. The biopsy was sent to the lab (figure 1B).
The results showed tumorous tissue composed of rounded and spindly cells with atypical nuclei and increased mitosis, these cells were found to be positive for myogenin and desmin (positive), while negative for S100 and CD 68 (Figure 3). The findings were consistent with embryonal rhabdomyosarcoma.
After the patient’s recovery, he was managed by the oncology team.
Figure 1, 2, 3, 4
Discussion
RMS is a form of a malignant soft tissue tumor that develops from either striated muscle cells or mesenchymal cells that have differentiated from striated muscle cells. It accounts for approximately 6.5% of all malignancies in children under the age of 15, making it the most prevalent soft tissue sarcoma in children. However, it is extremely rare in adults [4]. Paratesticular RMS accounts for approximately 7% of genitourinary RMS cases [9].
Only 15–25% of RMS cases are reported to originate from the prostate, bladder, testicular envelopes, and epididymis. This includes the following histological: anaplastic, spindle cell embryonal, alveolar, and embryonal-botryoid [11]. Embryonal RMS (eRMS), which accounts for 60% of cases, is the most common subtype. [5].
Paediatric RMS patients are treated with a multimodality strategy that combines radiation treatment, surgery, and/or induction (multidrug) chemotherapy. The Intergroup Rhabdomyosarcoma Study Group (IRSG) reports that this multimodality treatment has improved the overall survival (OS) at five years for young patients with localised disease. However, individuals with metastatic disease still have a poor survival rate (26%–27% vs. 66%–85%) [6]. This implies that early diagnosis is crucial for improving the prognosis of paediatric RMS patients.
Typical patient presentations with paratesticular RMS include a painless scrotal lump or symptoms of metastasis like fatigue, loss of appetite, weight loss, and inguinal lymphadenopathy. Pain is noted in only 7% of the instances [8]. However, in our case, the scrotal swelling was thought to be a consequence of the direct trauma that our patient had.
The initial imaging of choice is scrotal sonography. However, paratesticular rhabdomyosarcoma exhibits nonspecific sonographic features. It is critical to diagnose an intrascrotal extra testicular tumor with mixed echogenicity and hypervascularity [9], as seen in our case. In the differential diagnosis, consideration should be given to adenomatous tumors, leiomyomata, and other solid extratesticular masses. Direct invasion of the testicular tunica by hematogenous or lymphatic pathways is a common route for metastases [10]. The most locations for metastasis are the lungs, cortical bone, and local lymphatic systems [10].
MRI and computed tomography may both be used to determine the location, size, and presence of distant metastases in a tumor. [13] MRI scans can reveal a heterogeneously enhancing mass within the testis or a mass compressing the testis in the paratesticular region [13]. PET/CT scans can offer precise information regarding cancer metastases. However, none of these is a confirming procedure. For a definite diagnosis of RMS, a histopathologic investigation is necessary.
The histological appearance of embryonal RMS is characterized by an undifferentiated patternless spindled cells and tiny round blue cells. Immunohistochemical markers are required to distinguish RMS from other primary mesenchymal and germ cell cancers with rhabdomyoblastic differentiation [14]. Since smooth muscle markers are not very useful in differential diagnosis, myo-D1 and myoglobin are more specific than muscle-specific action, smooth muscle actin, and desmin [19,20] ( Myoglobin was used in the diagnosis of our case)
A multimodal approach is required for the treatment of embryonal RMS. Surgical intervention alone yields a 50% two-year relapse-free survival rate prior to successful treatment [17]. Surgical treatment is preferably performed through high dissection radical orchiectomy and spermatic cord ligation via an inguinal incision, as in our case, not scrotal approach because of the risk of minute residual disease contamination. Although the scrotal method is used in up to 25% of cases [18].
All individuals with paratesticular RMS should undergo evaluation for additional treatment. Protocols involving doxorubicin and ifosfamide are available, along with the use of vincristine, actinomycin-D, cyclophosphamide, and cisplatin. According to the Minimal Clinical Recommendations and the European Society for Medical Oncology, doxorubicin is the first-line treatment for soft tissue sarcoma. There is no conclusive evidence That polychemotherapy improves overall survival compared to doxorubicin alone. However, when ifosfamide and doxorubicin are used together, the response rate is higher [21,22]. For all patients with microscopic residual disease and histopathologically positive lymph nodes, local radiotherapy is recommended in addition to systemic treatment [24]. Age is often associated with a poor prognosis, especially for patients over 10 years old or younger than 1 year old [16].
Conclusion
Because most patients are discovered inadvertently (as in our case), paratesticular RMS should be considered as part of during the routine diagnosis.
Conflict of Interests
There is no conflict of interest regarding the publication of this paper.
Acknowledgments
The authors would like to express their deepest appreciation to dr. Thaher Salman MD- Consultant
pathologist who reviewed the surgical specimen.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author.
Funding Statement
This research is Self-funding by the authors.
References
1. Parham DM, Barr FG. Classifcation of rhabdomyosarcoma and its molecular basis. Adv Anat Pathol. 2013; 20:387–97.
2. Rudzinski ER, Anderson JR, Hawkins DS, Skapek SX, Parham DM, Teot LA. The World Health Organization classifcation of skeletal muscle tumors in pediatric rhabdomyosarcoma: a report from the Children’s Oncology Group. Arch Pathol Lab Med. 2015;139:1281–7.
3. Weiss AR, Lyden ER, Anderson JR, et al.: Histologic and clinical characteristics can guide staging evaluations for children and adolescents with rhabdomyosarcoma: a report from the Children's Oncology Group Soft Tissue Sarcoma Committee. J Clin Oncol. 2013, 31:3226-32.
4. Ahmed HU, Arya M, Muneer A, Mushtaq I and Sebire NJ: Testicular and paratesticular tumours in the prepubertal population. Lancet Onco.l 2010,11: 476-483, 2010.
5. Qualman S, Lynch J, Bridge J, et al. Prevalence and clinical impact of anaplasia in childhood rhabdomyosarcoma: a report from the Soft Tissue Sarcoma Committee of the Children’s Oncology Group. Cancer 2008; 113:3242–7.
6. Raney B, Huh W, Hawkins D, Hayes-Jordan A, Million L, Rodeberg D, Teot L, Anderson J, Soft Tissue Sarcoma Committee of the Children’s Oncology Group A, CA. Outcome of patients with localized orbital sarcoma who relapsed following treatment on Intergroup Rhabdomyosarcoma Study Group (IRSG) Protocols-III and -IV, 1984–1997: a report from the Children’s Oncology Group. Pediatr Blood Cancer. 2013; 60:371–6.
7. Embryonal rhabdomyosarcoma of the epididymis presenting as epididymitis: A case report HONG-LIANG WANG1, LING-YUN LIU1, RUN-HUI TIAN2, FU-BIAO LI1 and KAI-MIN GUO. MOLECULAR AND CLINICAL ONCOLOGY. 2016, 4: 625-627.
8. Khoubehi B, Mishra V, Ali M, Motiwala H and Karim O: Adult paratesticular tumours. BJU Int.2002, 90: 707 715.
9. Mak CW, Chou CK, Su CC, Huan SK, Chang JM. Ultrasound diagnosis of paratesticular rhabdomyosarcoma. Br J Radiol 2004; 77:250-2.
10. Akbar SA, Sayyed TA, Jafri SZ, Hasteh F, Neill JS. Multimodality imaging of paratesticular neoplasms and their rare mimics. Radiographics 2003; 23:1461-76.
11. Burnette JO, Klaassen Z, Hatley RM, et al. Staging paratesticular rhabdomyosarcoma in the “as low as reasonably achievable” age: the case for PET-CT. Urology 2013;82:220–3.
12. Damazio E, Caran E, Ortiz V, et al. Does negative retroperitoneal CT in adolescents with paratesticular rhabdomyosarcoma preclude the need of retroperitoneal lymph node dissection? Einstein 2011; 9:527–9.
13. Mason BJ, Kier R. Sonographic and MR imaging appearances of paratesticular rhabdomyosarcoma. AJR Am J Roentgenol 1998; 171:523–4.
14. Kahn DG: Rhabdomyosarcoma mimicking acute leukemia in an adult: Report of a case with histologic, flow cytometric, cytogenetic, immunohistochemical, and ultrastructural studies. Arch Pathol Lab Med .1998;122: 375-378.
15. Hardaway CA, Graham BS, Barnette DJ and Feldman BD: Embryonal rhabdomyosarcoma presenting in an adult: A case report and discussion of immunohistochemical staining. Am J Dermatopathol .2003;25: 45 52.
16. Radiol. 1995, 50:130-1. 10.1016/s0009-9260(05)82999-x 15. Venyo AK: Rhabdomyosarcoma of the testis, epididymis and spermatic cord: a review and u pdate. Pulsus J Surg Res. 2018; 2:78-86.
17. Sutow WW, Sullivan MP, Ried HL, Taylor HG, Griffith KM: Prognosis in childhood rhabdomyosarcoma. Cancer. 1970; 25:1384-90.
18. Cecchetto G, De Corti F, Rogers T, Martelli H: Surgical compliance with guidelines for paratesticular rhabdomyosarcoma (RMS). Data from the European Study on non-metastatic RMS. J Pediatr Surg. 2012; 47:2161-2.
19. Dall'Igna P, Bisogno G, Ferrari A, et al.: Primary transcrotal excision for paratesticular rhabdomyosarcoma is hemiscrotectomy really mandatory. Cancer. 2003, 97:1981-4.
20. Rogers DA, Rao BN, Meyer WH, Pappo A, Lobe TE, Fleming ID, Kauffman WM: Indications for hemiscrotectomy in the management of genitourinary tumors in children. J Pediatr Surg. 1995, 30:1437-9.
21. Von Mehren M, Benjamin RS, Bui MM, et al.: Soft tissue sarcoma, version 2.2012: featured updates to the NCCN guidelines. Natl Compr Canc Netw. 2012, 10:951-60.
22. Casali PG, Blay JY: Soft tissue sarcomas: ESMO clinical practice guidelines for diagnosis, treatment and 2022 Al Ghamdi et al. Cureus 14(5): e24786. DOI 10.7759/cureus.24786 6 of 7 follow-up. Ann Oncol. 2010, 21 Suppl 5: v198-203..
23. Ferrari A, Casanova M, Massimino M, Luksch R, Piva L, Fossati-Bellani F: The management of paratesticular rhabdomyosarcoma: a single institutional experience with 44 consecutive children. J Urol. 1998, 159:1031-4.
24. Ogilvie CM, Crawford EA, Slotcavage RL, King JJ, Lackman RD, Hartner L, Staddon AP: Treatment of adult rhabdomyosarcoma. Am J Clin Oncol. 2010, 33:128-31.
25. Faure, iakit , anait , Chaumo tre , Rome A, Merrot T. Le rhabdo- myosarcome paratesticulaire de l’enfant : une urgence scrotale. rch Pediatr, 2012; 19: 1340–4.
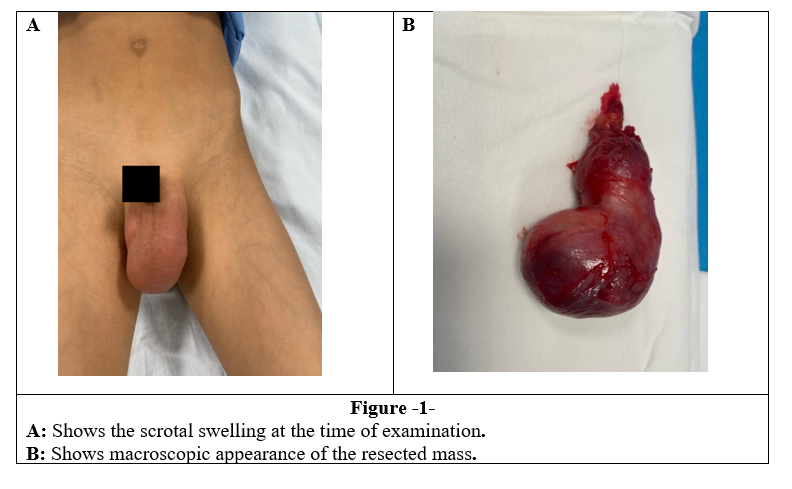
Figure 1
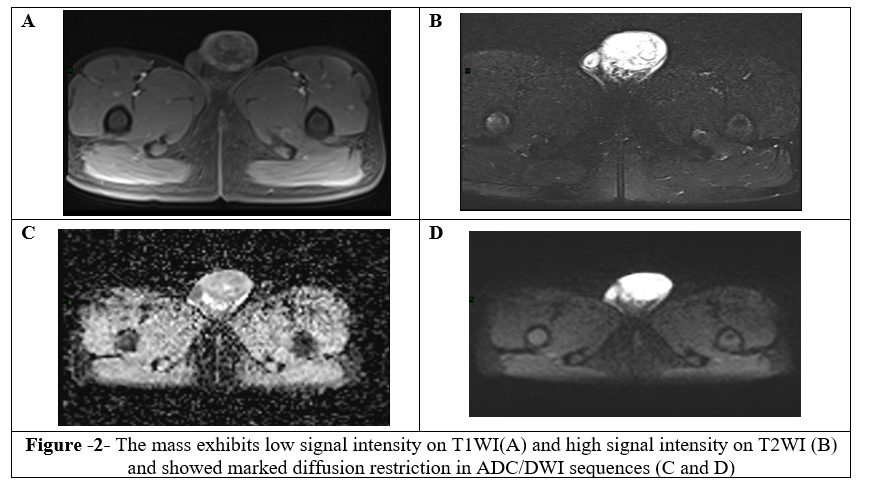
Figure 2
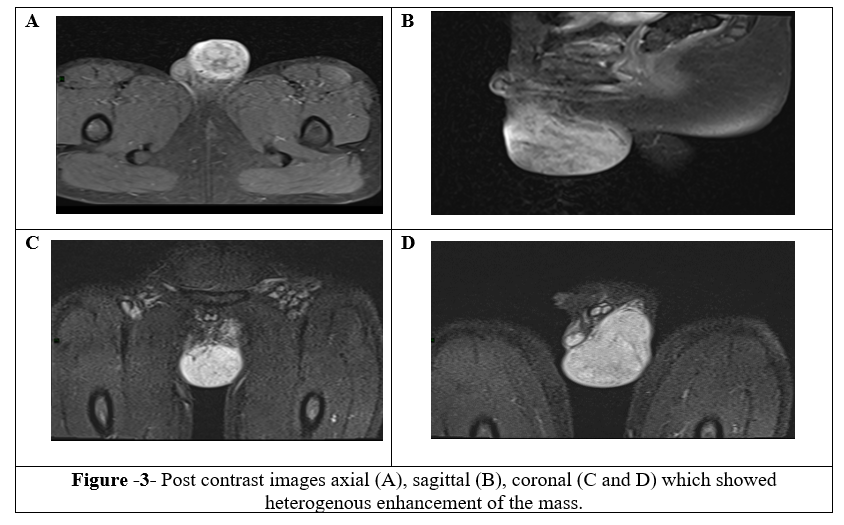
Figure 3
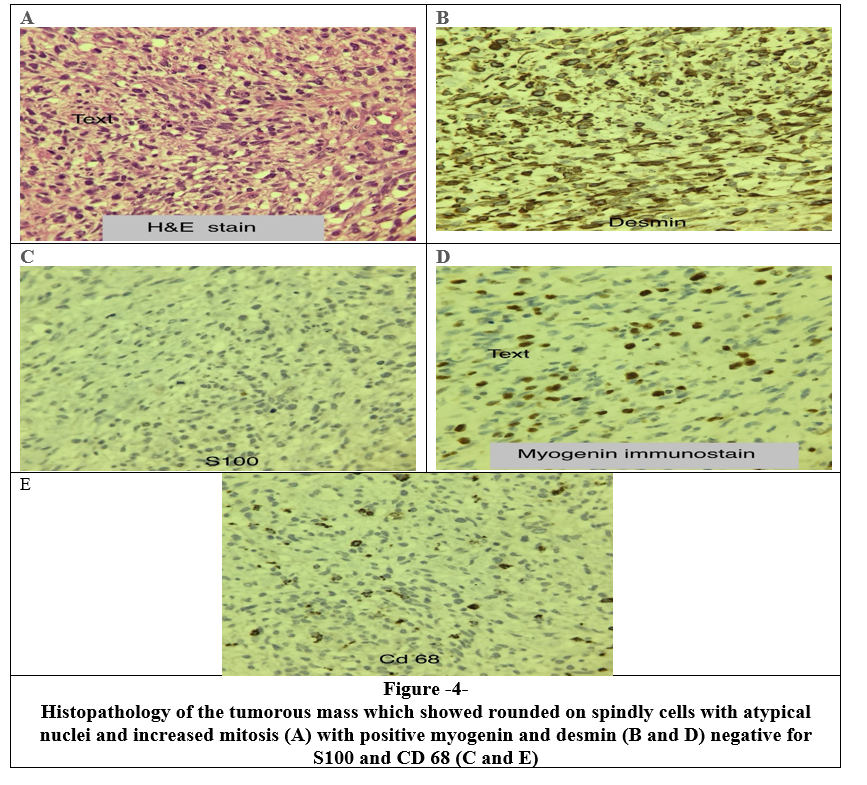
Figure 4