Errors Innate of the Metabolism
Errors Innate of the Metabolism
Drs. Patricia Aguero1, Veronica Gonzalez2, Aida Lemes3, Isabel Pigola4, Rosa Finozzi5, Beatriz Mendoza6.
1. Patricia Omen - Medical Specialist in Endocrinology and Metabolism. Assistant of Clinic of Endocrinology and Metabolism UdelaR. Medical of polyclinic of CRENADECER. Institute of the Security Social B.P.S.
2. Student of PROINBIO Master's Degree. Pediatrician, former resident CRENADECER Inborn Errors of Metabolism Polyclinic. Security Institute Social B.P.S.
3. Genetics medical. Polyclinic of Errors Innate of the Metabolism CRENADECER. Institute of the Security Social B.P.S.
4. Resident 1 year, Endocrinology and Metabolism Clinic UdelaR. Mariana Painted -Medical Specialist in Endocrinology and Metabolism. Teaching collaborator Service of Endocrinology Pediatric UdelaR, CHPR.
5. Teacher Attached Clinic of Endocrinology and Metabolism UdelaR. Coordinator of the Service of Endocrinology Pediatric UdelaR-CHPR Medical holder Service of Endocrinology pediatric ASSE.
6. Professor at the UdelaR Endocrinology and Metabolism Clinic. Director of the Service of Endocrinology Pediatric UdelaR, CHPR. Medical Endocrinologist holder of CRENADECER.
*Correspondence to: Dr. Rosa Finozzi, Teacher Attached Clinic of Endocrinology and Metabolism UdelaR. Coordinator of the Service of Endocrinology Pediatric UdelaR-CHPR Medical holder Service of Endocrinology pediatric ASSE.
Copyright
© 2024 Dr. Rosa Finozzi. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 17 Aug 2024
Published: 10 Sep 2024
Errors Innate of the Metabolism
Aim
Generate tools for clinical suspicion, diagnosis and correct treatment of the inborn errors of metabolism (EIM) that occur in the neonatal period. In particular those that HE present as a disease serious that requires treatment of emergency he which can save them the life as well as avoiding neurological complications permanent.
Introduction
EIMs, also called “inherited metabolic disorders” 1 , cover a broad group of alterations biochemical of origin genetic in the structure either in the function of a protein 2 .
They can manifest at any age, both in pediatric age and in adolescence or adulthood 3 . In this chapter we will refer to those of neonatal presentation, and within them we will emphasis in those in the which a treatment prompt can change his forecast.
The suspicion of the presence of a EIM in the newborn may arise from history familiar, by he antecedent of a brother deceased with chart similar either Yeah there is parental consanguinity. But since the majority of cases are autosomal inheritance recessive does not always raise this alert in the interrogation. The immaturity of the newborn determines that have a very limited capacity for signs and symptoms in response to the most of the attacks. By this reason the presentation clinic is to slight nonspecific with signs and symptoms common with other more common diseases. Is Therefore, it is essential to include EIM within the differential diagnoses of the newborn. born sick and consider them in parallel to other more frequent pathologies 4 .
The Neonatal Screening Programs (PPN) contemplate the diagnosis of some EIM that have treatment specific able of change the natural history of the disease. In Uruguay in he PPN, this including he study of shape mandatory of Phenylketonuria (PKU) and Medium Chain Acyl Coenzyme A Dehydrogenase Deficiency (MCAD). The investigation The expanded version has been carried out on a pilot basis since 2008 in Uruguay. This is done through of tandem mass spectrometry (MS/MS) that can detect more than 20 disorders metabolic 2.
The increasing complexity of biochemical processes and molecular tests identified that accompany the progress technological current, do difficult to keep up with field of EIM, so creating critical thinking based on pathophysiology is help for doctors who are on the front lines of care. The idea is that the Clinical diagnosis of EIMs is based on a small number of important principles: which are detailed later.
Epidemiology
The EIM HE find inside of the diseases rare defined by a prevalence less than 1 case per 2,000 individuals. Since 1450 disorders are currently known hereditary metabolic disorders, it is estimated that their global incidence may be of up to 1/500 1.6 .
Etiology and pathophysiology
The EIM HE characterize by the accumulation or deficiency of a specific metabolite, caused usually due to an enzymatic defect 4
A classic classification of EIMs is the following:
Group I: alterations of intermediate metabolism. There is acute poisoning or chronic due to accumulation of compounds prior to the point of metabolic blockage. In this cluster They include disorders in the metabolism of amino acids such as PKU, maple syrup-smelling urine (MSUD) and homocystinuria, blood disorders metabolism of the carbohydrates such as galactosemia, blood cycle defects the urea, as well as organic acidurias and the porphyrias, among another 4 .
Group II: alterations in energy metabolism. They usually manifest clinically with symptoms caused by deficiency in the production either he use of energy in muscle, liver, myocardium or brain such as hypoglycemia, hyperlactatemia, acidosis, failure cardiac, etc Includes the defects mitochondrial that are in great extent intractable 4 .
Cluster III: alterations of the synthesis, prosecution or catabolism of molecules complex. The symptoms are generally permanent and inexorably progressive, independent of intercurrent events such as infections and not related to the type of food eaten. Includes EIMs in the synthesis of cholesterol, triglycerides, phospholipids, bile acids, purines and pyrimidines 4 .
Approach diagnostic
Clinical suspicion:
Some data of the anamnesis they must alert always about the possibility of be facing a patient with an EIM:
- Siblings who died due to a doubtful cause, sudden death or with a diagnosis of sepsis.
- Consanguinity between the parents 4 .
- Cholestasis gravidarum, syndrome of HELLP and liver fatty maternal during he pregnancy 4
- Be forehead to a R.N. previously healthy that presents acutely with symptoms serious 4 .
EIMs manifest in a varied and non-specific way and can affect any device. either system given the large number of defects possible enzymes 6 .
A high level of suspicion should be maintained in patients with acid-base status disorders. unexplained, hyperammonemia, hypoglycemia, hematological alterations, liver dysfunction and kidney disease 6 .
The onset and severity of symptoms may be affected by intercurrent infections, stress, fasting or eating certain foods 6.
Clinical presentation Prenatal
The EIM of start prenatal HE they can classify in 3 categories clinics main and These are generally disorders in which the prognosis cannot be modified in any way. significant, the majority correspond to groups II and III 4 .
Clinical categories:
1. Malformations older that They can present as skeletal malformations, heart disease congenital and neural tube defects 4 .
2. Dysplasias including heterotopias and/or cortical cysts, anomalies of the pit later, polycystic kidney disease and liver cysts 4 .
3. Alterations functional as delay of the growth intrauterine, dropsy fetal, liver disease, splenomegaly, microcephaly, or facial dysmorphism 4 .
Neonatal
They may present with nonspecific symptoms such as respiratory difficulty, apnea, hypotonia, refusal to feed, poor sucking reflex, vomiting, dehydration, hypoglycemia, compromised consciousness and seizures, problems that can easily be attributed to sepsis or other etiologies 4 .
In the conditions in which there is intoxication due to accumulation of a substrate (group I) Typically, this is a full-term newborn who, after pregnancy and birth normal, has a variable initial symptom-free period (from hours to weeks) and then deteriorates quickly without reason apparent and not responds to the symptomatic therapy 4 .
In shape schematic and simplified, they can differentiate 4 patterns clinical 8 :
1) Neurological
2) Hypoglycemia
3) Hepatic
4) Cardiac
1) Neurological: is the presentation further common (hypotonia and encephalopathy: lethargy, hyporeactivity, comma) 8.9 .
Urea cycle defects present with neurological impairment, hyperammonemia, and Initial respiratory alkalosis that, as the patient worsens, can progress to metabolic acidosis. Consider initial respiratory alkalosis is key to diagnostic orientation 4 .
The acidemias organic also HE associate deterioration neurological but with acidosis metabolic and remaining anion increased, there may or may not be increase in ammonia 9 .
Nonketotic hyperglycinemia may present with seizures that are difficult to control with a pattern EEG shock-suppression and hypotonia 9 .
In case of seizures that do not respond to anticonvulsant therapy, the sensitive vitamins: pyridoxine, pyridoxal phosphate, folic acid and biotin, due to the possibility of treatment specific 8 .
Energy deficiency diseases (group II) do not present asymptomatic periods and they usually manifest with hypotonia widespread, seizures and deterioration neurological rapidly progressive. The newborn may present dysmorphisms or malformations. The acidosis Primary lactic acid is a biochemical manifestation of this group 8.
2) Hypoglycemia: the levels of glycemia low in blood they must be corrected immediately. The 3 groups of diseases linked with hypoglycemia are: hyperinsulinism, glycogen storage diseases (GSD) and defects hereditary of beta-oxidation mitochondrial acids fatty (FAO) 8.
3) Hepatic: The hepatomegaly and cholestasis can be the manifestation initial of the galactosemia or tyrosinosis type I in a newborn with mobilization of transaminases, including or not a slight increase in lactic acid 4.
4) Heart failure: in neonates the treatable EIM diseases that can occur with heart failure include inherited defects of mitochondrial beta-oxidation of acids fatty (FAO), the disease of Pompe and mutation in TMEM70 8.
Diagnostic approach
When there is clinical suspicion of an EIM, we must perform basic common laboratory studies for confirm the existence of patterns biochemicals compatible with he disorder metabolic.
It is essential to take a critical sample immediately at the onset of symptoms or at moment of the decompensation already that can contribute information essential. With he treatment symptomatic, he patient can improve to the be eliminated the compounds abnormal accumulated either No reply to the same and die and HE lose so the chance diagnostic 12 .
A simple metabolic profile is within reach of any center and those samples that require a laboratory specialized they can be stored of shape correct until their analysis as shown in the following table.
Table 1
Board 1- Research protocol and storage of samples for the emergency 1,7 .
The studies complementary they will be guided by the assessment in set of the clinical presentation and initial basic biochemical profile, which may require an acid study organic in urine, amino acids of blood I in CSF, neurotransmitters in CSF, porphyrins in urine etc 7 . Others studies that they can contribute information valuable, are: chest x-ray, electrocardiogram, echocardiogram, electroencephalogram, ultrasound transfontanellar and study of cerebrospinal fluid 1 .
A correct methodology aid to delimit the syndromes biochemicals (hypoglycemia, hyperammonemia, acidosis, ketosis, etc.), allowing the differential diagnosis to begin in these patients 12 .
The investigation neonatal is a tool fundamental to consider for the orientation diagnosis in the sick newborn. It is known that in those EIM of clinical onset very early, the result of the neonatal screening may not yet be available at the time of delivery. start of the symptoms. Without embargo in view of the suspicion of a EIM is essential communicate to promptly contact the BPS laboratory (0800-1767) to request the results and in case of It may be necessary to send a new sample taken during the acute episode of the disease.
Treatment
Neonatal EIMs can cause true medical emergencies. Your treatment It requires a multidisciplinary team and the etiological diagnosis should never be delayed. the general treatment measures 10 .
We will detail to continuation he treatment essential of emergency aimed at correcting the alterations clinical life-threatening biochemical conditions in the NB, generally linked to the diseases of group I and II 11.
Clinical stabilization: does not depend on the specific metabolic disorder, but is based on the resulting biochemical alterations, such as acidosis, hypoglycemia, or hyperammonemia that require immediate intervention 11.
These patients they must have a appropriate maintenance thermal, respiratory assistance, medium hemodynamic and infectious prophylaxis 11. Be alert to the following points:
1) Hydroelectrolyte control:
Dehydration is a common phenomenon in these patients, due to anorexia and vomiting. that accompany neurological deterioration, polypnea associated with acidosis and hyperdiuresis given by the abnormal excretion of some organic acids. This is an essential aspect in a RN with suspected EIM. Adequate hydration with non-hypoosmolar fluids is recommended. and with 5 or 10% glucose to prevent catabolism, hypovolemia and kidney failure. Is important avoid the overhydration that can contribute in some cases to the edema brain (urea or maple syrup cycle defect -MSUD-) 11,12 . Avoid using solution Lactated Ringer's.
Guidance guideline: First 24 hours of life: 60-80 ml/kg/day.
24-48 hours of life: 80-100 ml/kg/day.
From 72 hours of life: 100-120 ml/kg/day. From the 5th-7th day: 130-150 ml/kg/day.
2) Acid base balance:
In organic acidemias, intense metabolic acidosis (pH <7.15 or HCO3 <10 mmol/L mEq) can be partially corrected by the administration of bicarbonate. In these cases it is recommended to use the following formula:
?NaHCO3 = deficit x kg x 0.3 boluses 2-3 mEq/kg + 1/6 molar perfusion
When acidosis is moderate, it is better not to treat it specifically due to the risk of hypernatremia iatrogenic and because can be a useful marker of accumulation toxic compounds in the first hours of treatment. It can be reversed with measures initials of treatment 11,12 .
Depending on the EIM and the severity of the case, clearance measures may be required. substrate, endogenous or exogenous.
Endogenous purification measures
Once the patient is stabilized, the most important objective is to reverse the catabolic state and stimulate anabolism to achieve a reduction of the body's toxic products 11,12 . He metabolite major that HE accumulate and generates toxicity usually be derivative of the proteins therefore it must be done:
1) Suspension of protein intake for no more than 48 hours. Its reintroduction must be progressive. Start with 0.3-0.5g/kg/day.
2) Water and caloric intake. The calorie intake should ensure 25% more than needs for his age in situation normal: serum glucose to the 10% to 10 mg/kg/min or 5% glucose in congenital lactic acidosis. It may become necessary the insulin (0.05 IU/kg/h) Yeah glucose >200 mg/dl or glucosuria as a form of maintain anabolism.
3) The contribution of lipids can also be used as an energy source, as long as it is not a disorder of mitochondrial beta oxidation of fatty acids is suspected.
4) Initially, an intravenous supply is usually necessary but it must be rotated to the line. enteral as soon as possible, administering antiemetics if necessary.
5) In cases of hyperammonemia due to a defect in the urea cycle, the use of of ammonium chelators such as intravenous sodium benzoate (initial dose: 250 mg/kg) and products intermediate as the arginine either citrulline (dose initial: 200 mg/kg). No we count in he country with benzoate of sodium in prepared for use intravenous by it which must be administered by via enteral 11,12 .
Exogenous purification measures
For EIMs with acute metabolic toxicity such as urea cycle defects, some organic acidosis and MSUD, when the mentioned therapeutic measures are insufficient in the removal of the substratum toxic, can be necessary start measures of depuration exogenous such as hemodialysis or hemodiafiltration. Peritoneal dialysis is very little effective and is considered a temporary measure either for transferring the patient to a health center high complexity or prior to any of the most effective techniques.
Table 2- Administration of cofactors, vitamins and detoxifying substances 7 .
Follow-up
It is important to keep in mind that a multidisciplinary team is required for an approach comprehensive and to provide the best care depending on which EIM is diagnosed.
In Uruguay We have a National Reference Center for Congenital Defects and Diseases rare (CRENADECER) where HE can carry out counterreference for follow-up or treatment for these patients 7.
Figure 1- Algorithm for the EIM with possibility of treatment in the newly born 8 . HIE: encephalopathy hypoxic-ischemic. OXPHOS: phosphorylation oxidative, PC: pyruvate carboxylase, PDH: pyruvate dehydrogenase
|
Points to remember: |
|
-No reject a EIM by absence of background relatives given that his inheritance is mainly recessive |
|
-Keep a high level of suspicion of a EIM when the symptoms No they improve with he initial treatment and other more common etiologies are ruled out |
|
-Consider an EIM in idiopathic refractory epilepsy |
|
-Think about an EIM and not miss the diagnosis of treatable conditions |
|
-Take the sample criticism in he moment of the decompensation and store it although No HE can process at the moment. |
|
-Start he treatment general independent of the cause and in parallel to others treatments of most frequent pathologies. |
|
-Request results of the investigation at 0800-1767 |
|
-Then of confirmed he diagnosis and established he treatment of the patient, HE has to offer genetic counseling for the family |
References
1. Ferreira C, Rahman S, Keller M, Saudubray J. An International Classification of Inherited Metabolic Disorders (ICIMD). Journal of Inherited Metabolic Disease. 2021;44(1):164-177.
2. Queijo C, Lemes A. 25 Years of Newborn Screening in Uruguay. Journal of Inborn Errors of Metabolism & Screening. 2021;9
3. Cabello J, Giugliani R. Inborn Errors of Metabolism. Clínica Las medical journal Counts. 2015;26(4):483-486.
4. Saudubray J, García-Cazorla TO. inborn Errors of Metabolism Overview: Pathophysiology, Manifestations, Evaluation, and Management. Pediatric Clinics of North America. 2018;65(2):179-208.
5. Martín M, Legarda M, Dalmau J. Inborn errors of metabolism: approach diagnosis in Primary Care. Bulletin of the Pediatric Society of Asturias, Cantabria, Castile and Lion. 2007;47:111-115.
6. Palaces TO, Garcia EITHER, Garcia-Silva M. Diagnosis of the errors innate of the metabolism. An Pediatr Cont. 2008;6(6):347-352.
7. Errors congenital of the metabolism. In: Assandri AND, Casurriaga TO, You Pear V, Notejane M, Vázquez M, Zunino C. Pediatric Care. 9th ed. Montevideo: Office from the Book-FEFMUR; 2020: 629-634.
8. Saudubray J, Garcia J. Clinical approach to inborn Errors of Metabolism in Pediatrics. In: Saudubray J, Baumgartner M, Walter J. Inborn Metabolic Diseases. 6a ed. Berlin; 2016: 4-70
9. Lemes A. Inborn errors of metabolism. Pediatric Archives of Uruguay. 2003;74(1):33-36.
10. Touati G, Mochel F, Rabier D. Diagnostic Procedures. En: Saudubray J, Baumgartner M, Walter J. Inborn Metabolic Diseases. 6a ed. Berlin; 2016: 91-107.
11. Saudubray J, Sedel F. Hereditary metabolic diseases: generalities, groups clinical and algorithms diagnoses. In: Sanjurjo Q, Baldellou TO, Garcia J. Diagnosis and treatment of inherited metabolic diseases. 3rd ed. Madrid; 2010: 63-111.
12. Rebage V, Garcia J, Baldellou TO, Lopez J, Grief J. Errors congenital of the metabolism in he period neonatal. In: Sanjurjo Q, Baldellou TO, Garcia J. Diagnosis and treatment of the diseases metabolic hereditary. 3a ed. Madrid; 2010: 121-140.
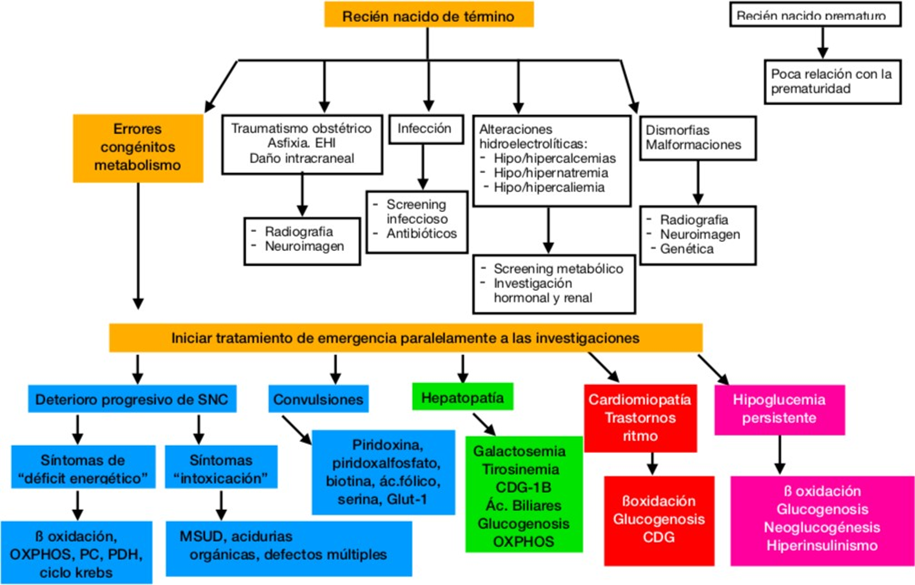
Figure 1
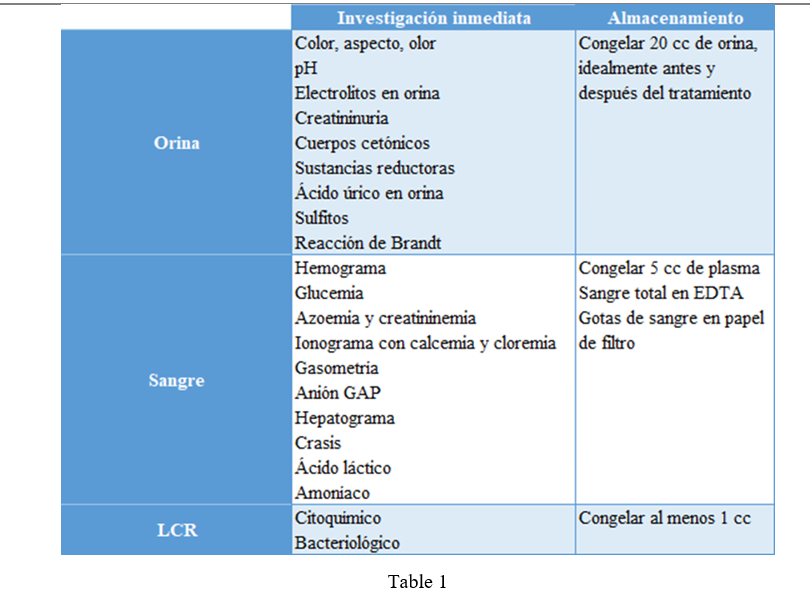
Figure 2
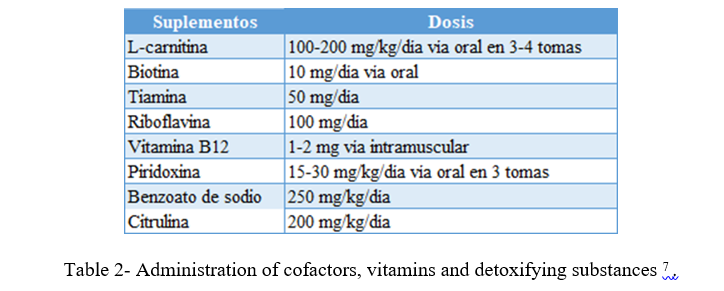
Figure 3
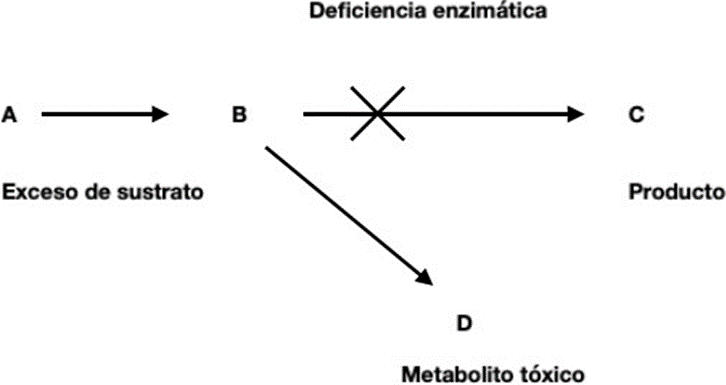
Figure 4