Challenges in the Management of "Autoimmune Polyglandular Syndrome Type I" during Critical Illness
Challenges in the Management of "Autoimmune Polyglandular Syndrome Type I" during Critical Illness
Juweria Shahrukh Effendi1, Muhammad Tahir Chohan1*, Samraiz Nafees1, Ali Javeed1, Javeria Hameed2, Sonia Zafar1, Fatima Ashfaq2 Komal Tahir1, Zulfiq ar Ahmed1, Muhammad Taqi1, Tadeusz Pawlak1
1. York and Scarborough Teaching Hospital NHS Foundation Trust, United Kingdom.
2. Nottingham University Hospital, United Kingdom.
*Correspondence to: Muhammad Tahir Chohan, York and Scarborough Teaching Hospital NHS Foundation Trust, United Kingdom. ORCID ID: 0009-0006-4620-8335.
Copyright
© 2025 Muhammad Tahir Chohan, This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 04 Aug 2025
Published: 16 Aug 2025
ABSTRACT:
Introduction: Autoimmune Polyglandular Syndrome Type I (APS1) is a rare monogenic disorder typically diagnosed in childhood. The clinical diagnosis requires at least two of the following features: hypoparathyroidism (HP), Addison’s disease (AD), and chronic mucocutaneous candidiasis [1]. Management involves lifelong hormone replacement, glucocorticoid and mineralocorticoid therapy for AD, and calcium with activated vitamin D for HP.
Case Presentation: We report the case of a young girl with a known diagnosis of APS1 since the age of five. She had remained relatively stable on a maintenance regimen of hydrocortisone, fludrocortisone, elemental calcium, and alfacalcidol. However, she presented with severe metabolic acidosis consistent with an Addisonian crisis (AC), precipitated by community acquired pneumonia and influenza A. Her clinical course was complicated by haemodynamic instability requiring vasopressor support and admission to the intensive therapy unit (ITU), as well as severe hypocalcaemia.
She was treated for sepsis and AC with intravenous antibiotics and high dose corticosteroids. Concomitant vulvovaginal and oral candidiasis were managed with oral fluconazole and topical Nystatin, respectively. During this period, she developed refractory hypocalcaemia necessitating repeated intravenous calcium infusions and significantly increased doses of elemental calcium and alfacalcidol.
Following the resolution of sepsis and a gradual tapering of corticosteroids, her calcium levels stabilised. This allowed a stepwise reduction of her calcium supplementation back to pre-admission levels: alfacalcidol 0.75 mg twice daily and elemental calcium 2 g/day, with sustained normocalcaemia.
Discussion: While patients with APS1 are generally stable on standard replacement therapy, acute intercurrent illness can significantly disrupt this equilibrium. In this case, systemic infection and high dose corticosteroid therapy contributed to severe and refractory hypocalcaemia, presenting a major management challenge. Careful monitoring and timely adjustment of calcium and activated vitamin D supplementation were essential to restore and maintain metabolic stability.
This case underscores the importance of anticipating metabolic decompensation and proactively managing calcium homeostasis in APS1 patients during periods of physiological stress.
Key Words: APS Type I: Autoimmune Polyglandular Syndrome Type I, CMC: chronic mucocutaneous candidiasis, Hypoparathyroidism, hypocalcemia, Endocrine Disorders, Autoimmune diseases.
Abbreviations:
APECED: Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy, AIRE: Autoimmune Regulator, CMC: chronic mucocutaneous candidiasis, PTH: parathyroid hormone, APS1: Autoimmune Polyglandular Syndrome Type I, AC: Addisonian crisis, ITU: intensive therapy unit, HCO3: Bicarbonate, CRP: C-Reactive protein, AD: Addison’s disease, HP: Hypoparathyroidism
Challenges in the Management of "Autoimmune Polyglandular Syndrome Type I" during Critical Illness
Introduction
Autoimmune Polyglandular Syndrome Type I, also termed as Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy (APECED), is an autosomal recessive disorder [2]. The mutation is in the AIRE (Autoimmune Regulator) gene, and clinically, the diagnostic criteria include the presence of at least two of its three major diagnostic features: chronic mucocutaneous candidiasis (CMC), hypoparathyroidism, and primary adrenal insufficiency (Addison’s disease).
It is reported that mostly the patients experience symptoms at an early age, nearly childhood, with CMC, which is considered the earliest manifestation, followed by endocrine dysfunction years later [1,3]. Unfortunately, the early manifestation of CMC is frequently reported to be resistant to standard antifungal treatment, primarily affecting the oral mucosa, nails, and skin.
Hypoparathyroidism is presented with signs and symptoms of hypocalcaemia, including paraesthesia, muscle cramps, tetany, and seizures. The biochemistry of these patients reports low serum calcium with low or undetectable parathyroid hormone (PTH) levels. Primary adrenal insufficiency is characterised by fatigue, hypotension, hyperpigmentation, hyponatremia, and hyperkalaemia, and may progress to life-threatening adrenal crisis if untreated [4].
Beyond these core features, APECED has a broad and heterogeneous clinical spectrum. Additional manifestations may include autoimmune thyroiditis, type 1 diabetes mellitus, autoimmune hepatitis, hypogonadism, asplenia, enamel hypoplasia, alopecia areata, and keratopathy. The clinical course is highly variable, even among individuals with the same AIRE mutation, and symptoms can present in a staggered manner over years or decades [5,6,7].
Although APECED is primarily a clinical diagnosis, genetic testing is increasingly employed to confirm AIRE mutations, especially in atypical presentations or populations with known founder mutations. Given its rarity, phenotypic diversity, and potential for delayed or incomplete expression, APECED remains a diagnostic and therapeutic challenge.
Case Presentation
A 21-year-old woman with a known diagnosis of Autoimmune Polyglandular Syndrome Type I (APS1) since the age of 5 had been relatively stable on maintenance therapy with hydrocortisone, fludrocortisone, elemental calcium, and alfacalcidol. She presented to the emergency department with a three-day history of generalised weakness, lethargy, productive cough, fever, nausea, vomiting, abdominal pain, and progressive drowsiness.
On admission, she was found to be hypotensive and hypoglycaemic. Blood gas analysis revealed severe metabolic acidosis consistent with an Addisonian crisis (AC). Work up identified community acquired pneumonia and influenza A as the precipitating factors.
She required intensive therapy unit (ITU) admission due to haemodynamic instability requiring vasopressor support, alongside profound hypocalcaemia.
She was managed successfully for sepsis, Addisonian crisis, and mucocutaneous candidiasis (vulvovaginal and oral) with intravenous antibiotics, intravenous hydrocortisone followed by oral steroids, and antifungal treatment with oral fluconazole and topical Nystatin.
However, her clinical course was complicated by recurrent and refractory hypocalcaemia, with serum calcium levels fluctuating between 1.40 and 2.10 mmol/L (reference range: 2.20–2.60 mmol/L). This required repeated intravenous calcium infusions and significant escalation of calcium supplementation, including elemental calcium up to 4.8 g/day and alfacalcidol up to 3 µg/day.
Following the resolution of sepsis and tapering of corticosteroid doses, her calcium levels gradually stabilised. This allowed for a stepwise reduction in supplementation back to her baseline maintenance doses: alfacalcidol 0.75 mg twice daily and elemental calcium 2 g/day, with sustained normocalcaemia thereafter.
Investigations:
|
Biochemistry |
Patient blood levels |
Reference Range |
|
WBCs |
12.9 |
4.0-11.0 |
|
Neutrophils |
8.97 |
2.00-8.00 |
|
CRP mg/L |
226 |
226 0-5 |
|
Urea (mmol/L) |
14.3 |
2.5-7.8 |
|
Creatinine (umol/L) |
107 |
45-84 |
|
Sodium (mmol/L) |
118 |
133-146 |
|
Potassium (mmol/L) |
5.0 |
3.5-5.3 |
|
Adjusted Calcium (mmol/L) |
2.07 |
2.20-2.60 |
|
PTH (pmol/L) |
<0.7 |
1.8-7.9 |
|
Serum Phosphate (mmol/L) |
1.48 |
0.80-1.50 |
|
Vitamin D (nmol/L) |
16 |
>51nmol/L |
|
Bicarbonate (mmol/L) |
14 |
22 – 29 |
|
Magnesium (mmol/L) |
0.67 |
0.7-1.1 |
|
Lipids, TSH, Amylase, B12 |
Normal |
|
|
Toxicology and Coeliac screen |
Negative |
|
Table 1: These are the patient's blood investigations throughout the hospital stay. WBC: white blood cell, CRP: C-Reactive protein, PTH: Parathyroid Hormone, HCO3: Bicarbonate.
Table 2: Shows the relationship between steroid, elemental calcium, and Alfacalcidol requirements during the hospital stay
Graph No.1: shows the Total serum calcium levels trend through the hospital stay
Graph No. 2: shows the CRP levels throughout the hospital stay.
Discussion
Patients with Autoimmune Polyglandular Syndrome Type I (APS1) generally remain clinically stable and asymptomatic while on standard maintenance therapy, which typically includes glucocorticoids, mineralocorticoids, calcium supplements, and activated vitamin D analogues such as alfacalcidol. However, this delicate metabolic balance can be significantly disrupted during episodes of acute illness, posing substantial management challenges. Several physiological stressors, including cytokine surges, impaired gastrointestinal absorption, increased metabolic demands, transient renal impairment, and resistance to activated vitamin D, can all contribute to disturbances in calcium homeostasis.
Graph number 1 shows our patient’s trend of serum calcium; hence, the development of profound and recurrent hypocalcaemia was multifactorial. The acute systemic inflammatory response to sepsis and influenza A infection, as observed in graph number 2 by visualising the significant rise of CRP at presentation on admission, likely led to a cytokine-mediated shift in calcium metabolism, resulting in reduced intestinal absorption and altered bone turnover. Additionally, the critical illness state increased metabolic demand and calcium utilisation, further exacerbating hypocalcaemia.
Compounding this was the administration of high-dose corticosteroids, which are necessary for the treatment of an Addisonian crisis. Corticosteroids are known to interfere with calcium balance through multiple mechanisms: they reduce intestinal calcium absorption by antagonising vitamin D action, increase renal calcium excretion, suppress bone resorption, and may lead to hypomagnesaemia, all of which contribute to secondary hypocalcaemia. Moreover, renal impairment during acute illness may have impaired the conversion and effectiveness of vitamin D analogues.
This complex interplay of factors resulted in resistance to conventional calcium and alfacalcidol therapy, necessitating significantly higher-than-usual doses and repeated intravenous calcium infusions to maintain biochemical stability. The eventual resolution of sepsis, along with tapering of corticosteroids, allowed for restoration of calcium balance and a return to baseline supplementation.
This case underscores the importance of anticipating metabolic decompensation in APS1 patients during acute illness and proactively adjusting calcium and vitamin D therapy. Close biochemical monitoring, early recognition of contributing factors, and a multidisciplinary approach are critical in avoiding life-threatening complications associated with severe hypocalcaemia in this vulnerable patient population.
Conclusion
The temporal alignment between the resolution of hypocalcaemia and the improvement of sepsis, along with the tapering of corticosteroids as seen in Table 2, suggests that an earlier return to maintenance steroid doses, once clinically appropriate, may facilitate faster correction of hypocalcaemia in APS1 patients during acute illness. This observation underscores the importance of carefully balancing the need for stress-dose corticosteroids in adrenal crises with their known adverse effects on calcium metabolism and supports considering steroid de-escalation as soon as the patient is hemodynamically stable and the acute inflammatory response subsides.
References
1. Husebye ES, Anderson MS, Kämpe O. Autoimmune Polyendocrine Syndromes. N Engl J Med. 2018;378(12):1132-1141. doi:10.1056/NEJMra1713301
2. Perniola R, Fierabracci A, Falorni A. Autoimmune Addison's Disease as Part of the Autoimmune Polyglandular Syndrome Type 1: Historical Overview and Current Evidence. Front Immunol. 2021 Feb 26;12:606860. doi: 10.3389/fimmu.2021.606860. PMID: 33717087; PMCID: PMC7953157.
3. Bug?l? NM, Carsote M, Stoica LE, Albulescu DM, ?uculin? MJ, Preda SA, Boicea AR, Alexandru DO. New Approach to Addison Disease: Oral Manifestations Due to Endocrine Dysfunction and Comorbidity Burden. Diagnostics (Basel). 2022 Aug 28;12(9):2080. doi: 10.3390/diagnostics12092080. PMID: 36140482; PMCID: PMC9497746.
4. Ferre EMN, Rose SR, Rosenzweig SD, et al. Redefined Clinical Features and Diagnostic Criteria in APECED. JCI Insight. 2016;1(13):e88782. doi:10.1172/jci.insight.88782
5. Kisand K, Peterson P. Autoimmune Polyendocrinopathy Candidiasis Ectodermal Dystrophy. J Clin Immunol. 2021;41(5):1047-1060. doi:10.1007/s10875-021-00990-6
6. Oftedal BE, Wolff ASB, Bratland E, et al. Radioactive Labelling of AIRE and Mutation Detection in APECED. Clin Immunol. 2009;132(3):347-356. doi:10.1016/j.clim.2009.05.011
7. Fernández Miró M, Colom Comí C, Godoy Lorenzo R. Autoimmune polyendocrinopathy. Med Clin (Barc). 2021 Sep 10;157(5):241-246. English, Spanish. doi:10.1016/j.medcli.2021.02.004. Epub 2021 May 3. PMID: 33958142.

Figure 1

Figure 2
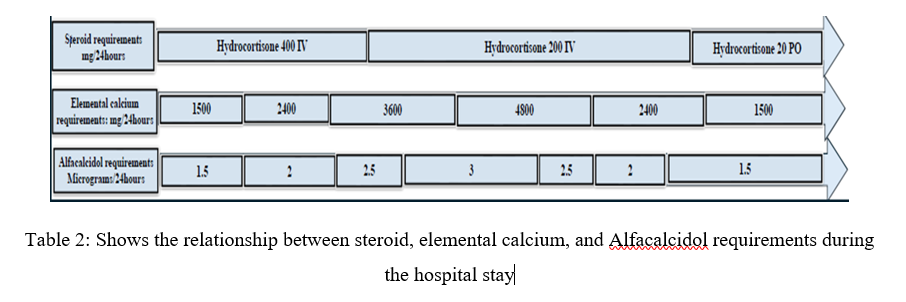
Figure 3