Aggressive Pancreatic ACTH-Secreting Neuroendocrine Tumor as a Rare Cause of Rapid-Onset Cushing’s Syndrome
Aggressive Pancreatic ACTH-Secreting Neuroendocrine Tumor as a Rare Cause of Rapid-Onset Cushing’s Syndrome
Oscar Paulo Reyes1, Ana Denise Sison2, Diana Colleen Dimayuga3, Michael Villa4
1,2,3,4. St. Luke’s Medical Center - Global City, Philippines.
*Correspondence to: Diana Colleen M. Dimayuga, MD, St. Luke’s Medical Center - Global City, Taguig, Metro Manila 1634, Philippines. ORCID https://orcid.org/0000-0002-0172-6941.
Copyright
© 2025 Diana Colleen M. Dimayuga, This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 25 Sep 2025
Published: 01 Oct 2025
DOI: https://doi.org/10.5281/zenodo.17283812
ABSTRACT:
Ectopic adrenocorticotropic hormone (ACTH) secretion is an uncommon cause of Cushing syndrome, with pancreatic neuroendocrine tumors (pACTHomas) being exceptionally rare. We report the case of a 31-year-old woman who presented with rapidly progressive Cushingoid features including facial rounding, acne, bruising, proximal muscle weakness, menstrual irregularity, and weight gain over six weeks. Laboratory tests showed severe hyperglycemia and hypokalemia, along with markedly elevated cortisol and ACTH. Imaging demonstrated a pancreatic mass consistent with a neuroendocrine tumor. Her clinical course was complicated by atrial fibrillation, stroke, and worsening metabolic disturbances. This case illustrates the aggressive nature of pACTHomas and the considerable challenges they present in both diagnosis and management.
Keywords: Ectopic ACTH syndrome; Pancreatic neuroendocrine tumor; Cushing’s syndrome; Refractory hypokalemia.
Aggressive Pancreatic ACTH-Secreting Neuroendocrine Tumor as a Rare Cause of Rapid-Onset Cushing’s Syndrome
Introduction
Ectopic ACTH secretion is an uncommon but serious cause of Cushing syndrome, most often originating from thoracic tumors and only rarely from pancreatic neuroendocrine tumors (pNETs).(1,2) When present, these tumors may cause fulminant hypercortisolism, resulting in profound metabolic disturbances, opportunistic infections, and high mortality if not promptly recognized.(1) Diagnosis is often difficult, as the clinical picture can mimic more common causes of Cushing syndrome, and is further complicated by the typically aggressive nature of the underlying tumor. Here, we report a case of fulminant Cushing syndrome caused by a large ACTH-secreting pNET.
Case Presentation
A 31-year-old woman with no prior medical history presented with 6 weeks of progressive facial and ankle swelling, 10-kg weight gain, acne, easy bruising, amenorrhea, and mood swings. On presentation, her blood pressure was 130/80 mm Hg, and body mass index was 28 kg/m². Physical examination revealed facial flushing, dorsocervical fat pad, violaceous striae, proximal muscle atrophy, and generalized hyperpigmentation. Neurologic and gynecologic examinations were unremarkable.
Laboratory results on admission showed a white blood cell count of 15,310/μL (reference, 6,000–10,000/μL) with 94% neutrophilia; hemoglobin, 16.4 g/dL (reference, 12–16 g/dL); hematocrit, 43.9% (reference, 37–47%); and platelets, 199,000/μL. The basic metabolic panel showed sodium, 140 mmol/L (reference, 135–145 mmol/L); potassium, 2.1 mmol/L (reference, 3.5–5.0 mmol/L); and creatinine, 0.86 mg/dL (reference, 0.7–1.2 mg/dL). Fasting blood glucose was elevated at 188 mg/dL (reference, 70–100 mg/dL), and hemoglobin A1c was 10% (reference, <6%). Thyroid-stimulating hormone was 0.122 μIU/mL (reference, 0.4–4.7 μIU/mL), free T4 was 0.58 ng/dL (reference, 0.89–1.76 ng/dL), and free T3 was 0.77 pg/mL (reference, 2.3–4.2 pg/mL). Liver function tests were within normal limits.
A low-dose 1-mg overnight dexamethasone suppression test produced an elevated serum cortisol of 991.03 nmol/L, showing unsuppressed morning cortisol, with an ACTH level of 577 pg/mL (reference, 7.2–63.3 pg/mL). Because of the markedly elevated ACTH, magnetic resonance imaging of the brain was performed to evaluate for a pituitary source but was negative (Figure 1). Cortisol levels remained elevated at >1750 nmol/L after the 8-mg high-dose dexamethasone suppression test, suggestive of an ectopic source.
Computed tomography (CT) of the chest revealed multiple non-calcified subcentimeter pulmonary nodules (Figure 2). CT of the abdomen demonstrated a 7.5 × 10 × 10.9 cm lobulated, heterogeneously enhancing mass in the pancreatic body and tail (Figure 3A), invading the gastric fundus and encasing the splenic artery (Figure 3B), worrisome for malignancy.
Endoscopic ultrasound–guided biopsy of the pancreatic mass confirmed a well-differentiated neuroendocrine tumor, positive for chromogranin, synaptophysin, and ACTH, establishing it as the source of ectopic ACTH production (Figure 4A–3C). Ki-67 proliferation index was 27%, consistent with a World Health Organization grade 3 neuroendocrine tumor with poor prognosis (Figure 4D). The hospital course was further complicated by refractory hypokalemia and severe hyperglycemia, both attributed to hypercortisolism.
Figure 1. Coronal view of a brain magnetic resonance imaging with contrast showing normal pituitary gland.
Figure 2: Axial view of chest computed tomography with contrast revealing subcentimeter calcified pulmonary nodules.
Figure 3A Axial view from abdominal computed tomography with contrast that revealed a large, lobulated, heterogeneously-enhancing, mass arising from the pancreatic body and tail is identified measuring 7.5cm x 10cm x 10.9cm.
Figure 3B Axial view of abdominal computed tomography showing the associated encasement of the splenic artery.
Figure 4 Immunohistochemistry result. (A) Synaptophysin: diffuse and strong positive staining. (B) Chromogranin: tumor cells are strongly positive. (C) ACTH: confirming differentiation. (D) the Ki-67 labeling index is at least 27%, indicating a high proliferative rate.
Treatment
The patient received aggressive potassium replacement and was initiated on insulin therapy. Surgery would have been the best option if the tumor was removable, but the situation was far more complicated. The mass was unusually large, pressing on the stomach (lesser curvature, body, and fundus) and the hilum of the spleen. It also encased the splenic artery, which made complete resection nearly impossible and raised serious concerns about the risk of major surgical complications.
Alternative options for controlling the hypercortisolism such as ketoconazole, metyrapone, etomidate, and chemotherapy were considered. However none were started as the patient declined further treatment.
Outcome and Follow-up
The patient was discharged and was able to return home; however, her condition continued to deteriorate. She ultimately succumbed to complications of multi-organ dysfunction before definitive management could be initiated.
Discussion
Ectopic ACTH syndrome (EAS) due to pancreatic neuroendocrine tumors (pACTHomas) is extremely uncommon, accounting for fewer than 10% of all EAS cases.(3,4) Compared with more frequent sources such as small-cell lung cancer or bronchial carcinoids, ACTH-producing pancreatic tumors often present late, with larger tumor burden, aggressive histology, and severe metabolic complications.(4,5)
This case stands out for several reasons. First, the patient’s age is striking. Most reported pACTHomas occur in individuals in their fourth to sixth decades of life;(4,5) our patient, at only 31 years old, is among the youngest described to date. Second, the pace of disease progression was extraordinary. The patient developed florid features of Cushing syndrome over just six weeks, a tempo that strongly suggests an ectopic ACTH source.(6) Third, the initial biochemical profile was remarkable for profound hyperglycemia, refractory hypokalemia, and suppression of thyroid function tests—all reflecting severe cortisol excess. Finally, the tumor was strikingly large and invasive with direct extension into the gastric wall and encasement of the splenic artery. Reported pACTHomas average around 4.4 cm in size,(4,5) highlighting just how aggressive our patient’s tumor was.
Management of such patients is challenging. Surgery is the management of choice when the tumor can be removed, but in this case the disease was too advanced for curative resection. Medical options such as ketoconazole, metyrapone, or etomidate could have been used to help control cortisol levels, but the patient declined therapy, which limited attempts to stabilize her rapidly worsening condition. Despite multidisciplinary evaluation, she succumbed to complications of multi-organ dysfunction shortly after discharge.
Although our patient passed away before definitive treatment could be given, this case is still worth reporting. The unusually young age at presentation, the rapid progression, and the severe biochemical abnormalities expand what is known about the clinical spectrum of this condition. The difficulties we faced including the tumor being unresectable, the urgent need for medical therapy to control cortisol, and the impact of the patient’s treatment decisions, offer important lessons for clinicians. Even when outcomes are poor, sharing these experiences helps raise awareness, encourages earlier recognition, and emphasizes the importance of timely multidisciplinary management in fulminant Cushing syndrome.
Learning Points
- Ectopic ACTH syndrome should be suspected in patients with rapidly progressive Cushing features and severe metabolic derangements.
- Profound and simultaneous metabolic disturbances (refractory hypokalemia, severe hyperglycemia, thyroid suppression) may indicate extreme cortisol excess.
- Large, invasive pancreatic NETs are often unresectable, highlighting the importance of early detection before vascular encasement occurs.
- Prompt initiation of cortisol-lowering therapy may be lifesaving when surgery is not feasible or delayed, as uncontrolled hypercortisolism drives morbidity and mortality.
Contributors
All authors made individual contributions to authorship. O.R., A.S., D.D., and M.V. were involved in the diagnosis and management of this patient. All authors reviewed and approved the final draft.
Funding: No public or commercial funding.
Disclosures: None declared.
Informed Patient Consent for Publication: Signed informed consent for publication was obtained directly from the patient.
Data Availability Statement: Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
References
1. Isidori AM, Lenzi A. Ectopic ACTH syndrome. Arq Bras Endocrinol Metabol. 2007;51(8):1217-1225.
2. Ilias I, Torpy DJ, Pacak K, Mullen N, Wesley RA, Nieman LK. Cushing’s syndrome due to ectopic corticotropin secretion: Twenty years’ experience at the NIH. J Clin Endocrinol Metab. 2005;90(8):4955-4962.
3. Alexandraki KI, Grossman AB. Ectopic ACTH syndrome. Front Horm Res. 2016;46:143-156.
4. Patel FB, Khagi S, Daly KP, Lechan RM, Ummaritchot V, Saif MW. Pancreatic neuroendocrine tumor with ectopic adrenocorticotropin production: a case report and review of literature. Anticancer Res. 2013 Sep;33(9):4001-5. PMID: 24023341.
5. Isidori AM, Kaltsas GA, Pozza C, et al. The ectopic adrenocorticotropin syndrome: clinical features, diagnosis, management, and long-term follow-up. J Clin Endocrinol Metab. 2006;91(2):371-377.
6. Davi MV, Cosaro E, Piacentini S, et al. Prognostic factors in ectopic Cushing’s syndrome due to neuroendocrine tumors: a multicenter study. Eur J Endocrinol. 2017;176(4):453-461.
7. al Saifi S, Druce I, Vickers M, Dennis K, A case report of bilateral adrenal gland stereotactic body radiotherapy to manage hypercortisolemia in a patient with ectopic adrenocorticotropic hormone (ACTH) production from a metastatic pancreatic neuroendocrine tumor: Cureus, 2024; 16(4); e57852
8. Shayesteh S, Fouladi DF, Fishman EK, Kawamoto S, Ectopic Cushing syndrome caused by a pancreatic neuroendocrine tumor: A case report: Radiol Case Rep, 2020; 15(7); 1014-17
9. Murakami M, Hirahata K, Fujimori N, Yamamoto T, Oda Y, Kozono S, Ueda K, Ito T, Nakamura M, Ogawa Y. Two cases of pancreatic neuroeFndocrine tumors with ectopic ACTH syndrome during their disease course. Clin J Gastroenterol. 2024 Apr;17(2):363-370. doi: 10.1007/s12328-023-01908-5. Epub 2024 Jan 20. PMID: 38244178.
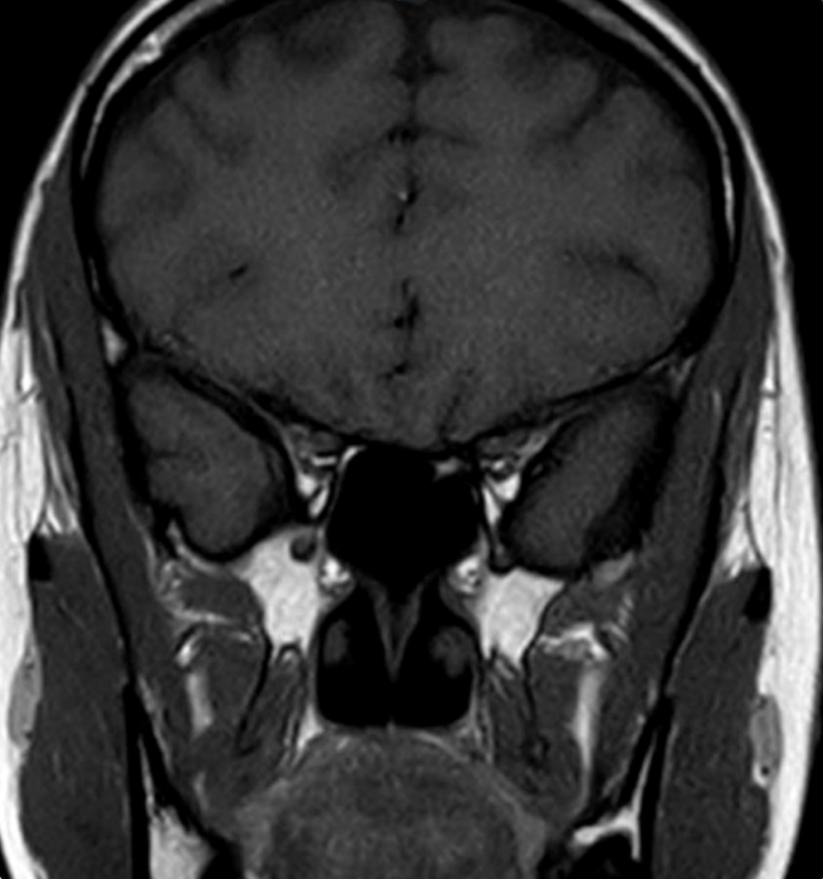
Figure 1
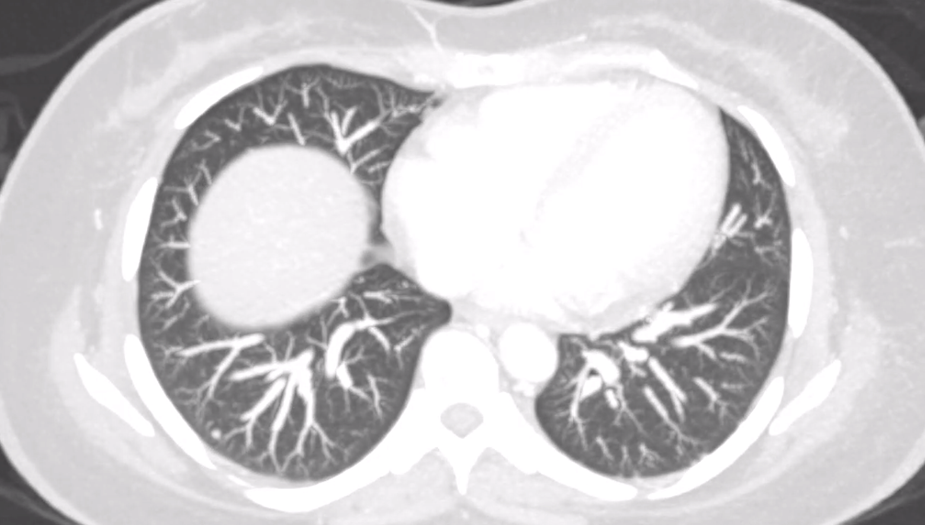
Figure 2
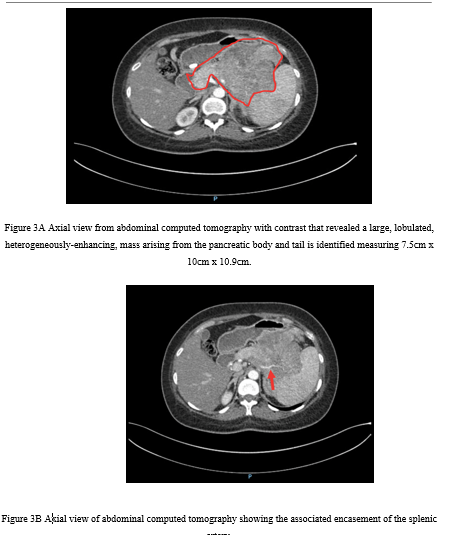
Figure 3
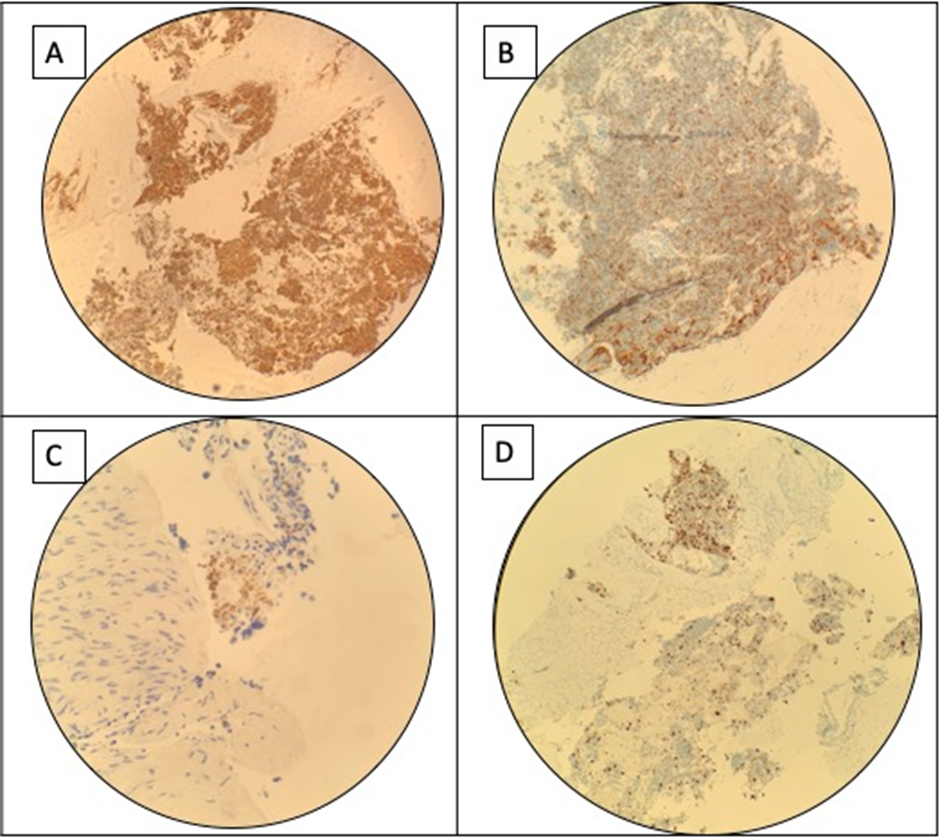
Figure 4