Recalcitrant Humeral Shaft Nonunion; A Case Report
Recalcitrant Humeral Shaft Nonunion; A Case Report
Dr. Talal H. Almalki 1, Abdullah Alsultan *1, Ziad Aljaafri 2, Mansour Alshehri 1, Ahmad Alajlan 1
1. Department of Orthopedic Surgery, Security Force Hospital, Riyadh, Saudi Arabia
2. College of Medicine, King Saud bin Abdulaziz University for Health Sciences, Riyadh, Saudi Arabia
*Correspondence to: Dr. Abdullah Alsultan, Security Forces Hospital Program Riyadh, Saudi Arabia.
Copyright
© 2023 Dr. Abdullah Alsultan. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 21 September 2023
Published: 30 September 2023
Abstract
Nonunion is defined as “established when a minimum of 9 months has elapsed since injury and the fracture shows no visible progressive signs of healing for 3 months”. The incidence of nonunion after operative treatment of humeral shaft fractures has been reported to range between 2.5% and 13%., we report the case of a 55-year-old woman with multiple comorbidities who presented to the emergency department at our institute 9 months post open reduction with internal fixation of the right humeral shaft after hearing a sudden pop associated with severe pain while performing daily activity with no significant trauma. Our case reveals how problematic humeral shaft nonunion can present, with our patient undergoing four revisions after the primary surgery to reach full radiological and clinical union, with an estimated period of 21 months after the last surgical revision. Functionality was fully restored, and no residual issues were encountered.
Keywords: Humeral shaft; Nonunion; Fractures; Trauma.
Recalcitrant Humeral Shaft Nonunion; A Case Report
Introduction
Nonunion was defined by the American Food and Drug Administration (FDA) in 1988 as “established when a minimum of 9 months has elapsed since injury and the fracture shows no visible progressive signs of healing for 3 months” [1]. It can be classified according to the vitality and healing potential of the fracture as hypervascular or avascular nonunion [2]. Humeral shaft fractures represent 3% of all managed fractures and occur with an incidence of 13 per 100,000 per year [3,4]. The incidence of nonunion after operative treatment of humeral shaft fractures has been reported to range between 2.5% and 13% [5-8].
Various methods have been reported for treating humeral shaft nonunion, but fewer are described for recalcitrant nonunion [9]. The purpose of this report is to present a possible option for treating recalcitrant humeral shaft fracture nonunion by presenting a case that required five procedures. Multiple modalities were used with limited success, and union was ultimately achieved by using a titanium mesh cage , reamer-irrigator-aspirator autograft, a long proximal humerus locking plate augmented with a 90 degrees 3.5 cm locking compression plate, and recombinant human bone morphogenetic protein-2 (rhBMP-2).
Case Presentation
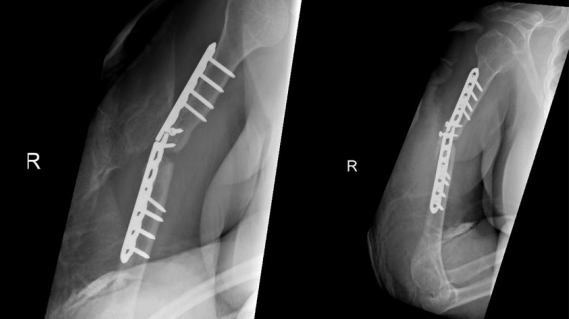
We present a case of recalcitrant humeral shaft nonunion in a 55-year-old obese female with type II diabetes, hypertension, and dyslipidemia. She sustained a rt humeral fracture in September 2017 and was treated at other facility with regular with 3.5 LC-DCP using 2 lag, 2 cortical, 6 locking , and 23 June 2018, she presented to the emergency after hearing a sudden pop associated with severe pain while performing daily activity with no significant trauma. Clinical examination revealed a healed surgical scar on the lateral aspect of the right arm, tenderness on palpation over the mid portion of the right arm, limitation of movement of the right shoulder and elbow due to pain, and intact distal pulses with no neurological deficits. Imaging in the form of X-rays revealed non-united right humeral shaft fracture. The fracture edges were smooth and partly sclerotic, with failure of a 3.5 mm locking compression plate was present in the form of plate fracture and screw pull-out at the level of the fracture (Figure 1). Laboratory investigations were negative for infection.
Figure 01
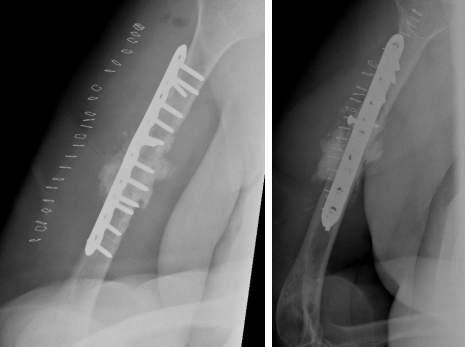
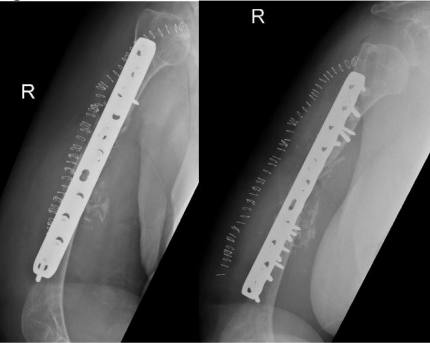
The patient was admitted and on 27 June 2018 underwent revision open reduction and internal fixation with the same surgical approach as the previous surgery. The fracture surfaces were trimmed until punctate bleeding was observed, and then the fracture was fixed with dynamic compression plating using a 10-hole 4.5 mm plate. An allograft was applied in the form of cancellous chips and putty bone substitute. Postoperative radiographs are shown in Figure 2.
Figure 2
On serial follow-ups, the patient was asymptomatic clinically and had no evidence of healing on radiographs. Then, on 15 May 2019, around 9 months post first revision she reported persistent localized pain to the fracture site, and with radiographic evidence of complete resorption of the allograft and non healed fracture a nonunion diagnosis was established (Figure 3).
Figure 03
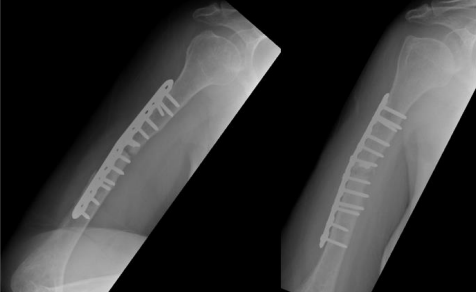
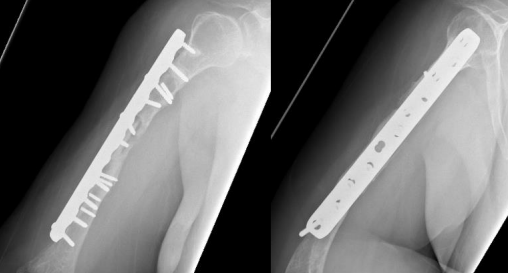
Options was discussed and The decision was made to take the patient for surgery for the third time “second revision” on17 June 2019. The sclerotic bone was excised, which accounted for 2.5 cm in length, and replaced with a tricortical iliac crest strut autograft, cancellous graft, and putty bone substitute. The same plate was applied, and new screws were used to hold the graft in place. The remaining screws were inserted, and the device was stable intraoperatively. Postoperative radiographs are shown in Figure 4.
Figure 4
One month postoperatively, on 29 July 2018 the patient heard a pop sound and felt sudden pain while performing physiotherapy. Examination revealed slight motion at the fracture site associated with pain. Radiographs showed four broken screws with displacement of the plate distally (Figure 5).
Figure 05
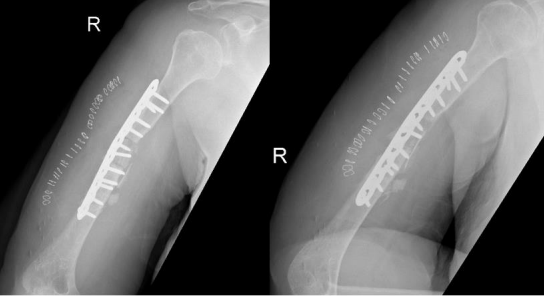
The patient was admitted and taken for her fourth surgery (third revision) on 1 August 2020. The implant and iliac crest bone graft were removed. Fibrous tissue was debrided, and fracture edges were freshened and medullary canals reopened. A 5 cm fibular autograft was harvested and placed in the bone defect, and fixation was performed with a broad 4.5 cm locking compression plate with cemented screws for proximal fixation. Postoperative radiographs are shown in Figure 6.
Figure 06
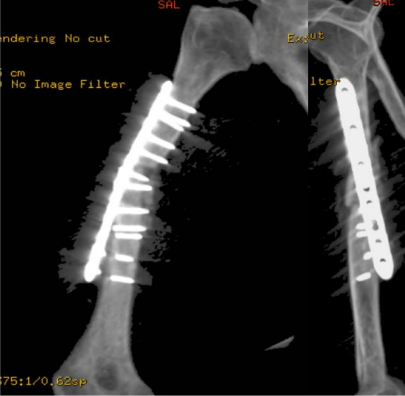
On serial follow-ups, the patient showed slow healing up to 6 months postoperatively. Follow ups continued for another 6 months but showed no progression of callus. Arrested partial healing was confirmed on CT scans, despite stable fixation, and the patient was symptomatic in the form of pain limiting daily activities. A bone scan was performed to evaluate for infection as the cause of partial healing and showed no evidence of infection. Radiographs at the 1-year follow-up are shown in Figure 7.
Figure 07
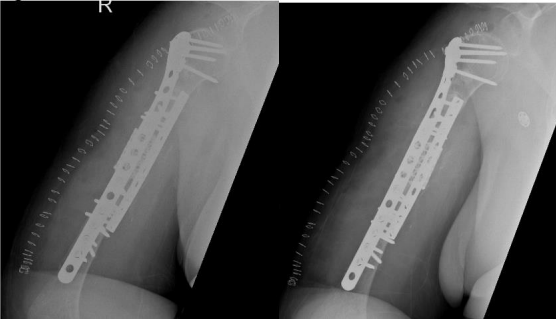
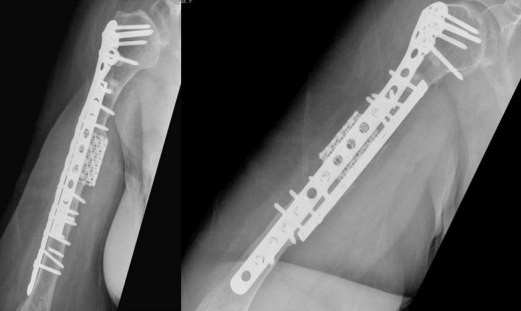
At that time, the primary surgeon decided to transfer the case to the Trauma unit, where the patient was reviewed thoroughly and all necessary investigations was made, after detailed discussion of the case and the options, we decided to go for another revision but with some tactical modifications. The patient was taken for yet another surgery, for the fifth time (fourth revision) on 30 November 2020. The same incision site was used, and the previous plate was removed and sclerotic bone debrided. An intramedullary cancellous bone graft was obtained from the patient’s left femur with the aid of a reamer-irrigator-aspirator. A titanium mesh cage was used to bridge the bone defect, and the graft from the left femur intramedullary canal was packed inside the titanium mesh cage. Primary fixation was obtained with a long proximal humerus locking plate laterally and augmented with a 3.5 cm locking compression plate anteriorly sat 90 degrees orientation.
Finally, 5.6 mL of rhBMP-2, on an absorbable collagen sponge carrier (ACS), was applied to the nonunion site. Postoperative radiographs are shown in Figure 8. Postoperatively, the patient suffered radial nerve neuropraxia, managed with splinting, and fully recovered 5 months postoperatively. Serial follow-ups for 21 months postoperatively showed complete healing clinically and radiographically, restoration of functional range of motion with the help of physiotherapy, and a painless functional limb. Radiographs are shown in Figure 9.
Figure 08
Figure 09
Discussion
Despite advancements in surgical techniques and equipment, cases of fracture nonunion are still encountered in clinical settings. Humeral shaft nonunion after failed surgical management can represent a challenge in achieving union and pain-free function. Various techniques have been described in the literature for managing recalcitrant humeral shaft nonunion [5,9-11], and multiple studies describe how humeral shaft nonunion can be managed with different approaches, including interlocking nail fixation, Ilizarov external fixation, and internal plate fixation with autologous iliac crest bone graft or vascularized fibular graft, which aims to provide adequate fixation across the fracture site and improve the local biomechanical environment and blood supply [12-14].
In our case, various methods were utilized before full radiological and clinical healing were achieved as multiple revisions were required. Applied methods included first trying to address nonunion with open reduction and internal fixation, as suggested by a recent study reporting a high rate of union following nonunion, even in recalcitrant nonunion, utilizing the same surgical approach from previous surgery [15]. However, this first revision surgery showed no evidence of radiological healing in our case.
In the second revision surgery, a tricortical iliac crest strut autograft, cancellous graft, and putty bone substitute were utilized. However, aggressive physiotherapy led to failure due to broken screws and plate displacement. Bone grafting has been shown to be effective in dealing with nonunion cases, with high success rates and fewer complications following the surgery [16]. The third revision was emergently performed with removal of the previous iliac crest bone graft. However, this time we applied a fibular autograft with locking compression plating and cemented screws. This method, utilizing compression plating and screws was found effective with or without grafting, and was proposed to facilitate bone healing, especially in younger patients [17]. However, in our case, slow healing followed by arrested healing was found during follow-ups.
Final revision surgery was performed with an intramedullary bone graft from the patient’s left femur, together with a titanium mesh cage to bridge the bone defect. The graft from the left femur intramedullary canal was packed inside the titanium mesh cage, and primary fixation was obtained with a long proximal humerus locking plate laterally, augmented with a 3.5 cm locking compression plate anteriorly. Finally, 5.6 mL of rhBMP-2 on an ACS was applied to the nonunion site.
Regarding titanium mesh cage utilization, one case is reported in which the patient had a defect of 8 cm following a gunshot. The patient was treated with external fixation for 4 months until soft tissue allowed for definite fixation, which was carried out utilizing a cylindrical titanium mesh cage that was packed with cancellous allograft and demineralized bone matrix and stabilized by compression plating. Union was achieved after 13 months, with no residual complications [18].
As described earlier for cases requiring multiple revisions, dual plating has been proved to improve the rate of healing following nonunion of humeral shaft fractures. Dual plating has multiple benefits, including axis correction, compression, and osteogenesis stimulation with grafting, all achievable during one surgery [19]. The success rate of achieving union with the use of dual compression plates in combination with autogenous grafting has been reported to be between 92% and 100% [20,21].
Utilization of rhBMP-2 has been proved effective in treating nonunion through its osteogenic and osteoconductive properties [22]. It has been described in multiple studies and proved to accelerate bone healing, which makes this a valid and noteworthy option when dealing with long bone nonunion cases [23]. In our case, rhBMP-2 was utilized in the final revision in combination with dual compression plating, and the patient achieved healing in a period of 5 months, which was in support of the literature.
The final revision surgery was the key surgery for reaching complete radiological and clinical healing, despite the presence of postsurgical radial nerve neuropraxia, which was managed with splinting, and the patient made a full recovery after 5 months. Restoration of functionality and range of motion was achieved with the aid of physiotherapy during the recovery period.
Conclusion
As shown in our case of how problematic humeral shaft nonunion can present, which necessitate in our opinion that such cases should be handled by a specialized trauma team. our patient underwent four revisions after the primary surgery to reach full radiological and clinical union, with an estimated period of 21 months after the last surgical revision. Functionality was fully restored, and no residual issues were encountered.
Reference
1. USFDA. Guidance Document for the Preparation of Investigational Device Exemptions and Premarket Approval Applications for Bone Growth Stimulator Devices. Rockvitte, MD: United States Food and Drug Administration; 1988.
2. Weber BG, Cech O. Pseudarthrosis, Pathology, Biomechanics, Therapy, Resutts. Bern: Hans Huber; 1976.
3. Court-Brown CM, Caesar B. Epidemiology of adult fractures: a review. Injury. 2006;37:691–697. doi: 10.1016/j.injury.2006.04.130.
4. Ekholm R, Adami J, Tidermark J, et al. Fractures of the shaft of the humerus. An epidemiological study of 401 fractures. J Bone Joint Surg Br. 2006;88:1469–1473. doi: 10.1302/0301-620X.88B11.17634.
5. Foster RJ, Dixon GL, Bach AW, Appleyard RW, Green TM. Internal fixation of fractures and nonunions of the humeral shaft. J Bone Joint Surg. (Am) 1985;67A:857–865.
6. Foulk DA, Szabo RM. Diaphyseal humerus fractures: Natural history and occurrence of nonunion. Orthopaedics. 1995;18:333–335. 10
7. Healy WL, White GM, Mick CA, Brooker AF, Weiland AJ. Nonunion of the humeral shaft. Clin Orthop. 1987;219:206–213.
8. Rosen H. The treatment of nonunions and pseudoarthrosis of the humeral shaft. Orthop Clin North America. 1990;21:725–742.
9. Borus TA, Yian EH, Karunakar MA. A case series and review of salvage surgery for refractory humeral shaft nonunion following two or more prior surgical procedures. Iowa Orthop J. 2005;25:194-9. PMID: 16089097; PMCID: PMC1888765.
10. Lin CL, Fang CK, Chiu FY, Chen CM, Chen TH. Revision with dynamic compression plate and cancellous bone graft for aseptic nonunion after surgical treatment of humeral shaft fracture. J Trauma. 2009 Dec;67(6):1393-6. Doi: 10.1097/TA.0b013e31818c1595. PMID: 20009693.
11. Feng, D., Wang, X., Sun, L. et al. Double plating with autogenous bone grafting as a salvage procedure for recalcitrant humeral shaft nonunion. BMC Musculoskelet Disord 21, 769 (2020). https:// www.ncbi.nlm.nih.gov/pmc/?term=humerus+shaft+fracture+nonunion
12. Leiblein M, Verboket R, Marzi I, et al. Nonunions of the humerus - treatment concepts and results of the last five years. Chin J Traumatol. 2019;22(4):187–195. doi: 10.1016/j.cjtee.2019.04.002.
13. Miska M, Findeisen S, Tanner M, et al. Treatment of nonunions in fractures of the humeral shaft according to the diamond concept. Bone Joint J. 2016;98-B(1):81–87. doi: 10.1302/0301-620X.98B1.35682.
14. Kashayi-Chowdojirao S, Vallurupalli A, Chilakamarri VK, et al. Role of autologous non-vascularised intramedullary fibular strut graft in humeral shaft nonunions following failed plating. J Clin Orthop Trauma. 2017;8(Suppl 2):S21–S30. doi: 10.1016/j.jcot.2016.12.006.
15. Oliver, William M. LLB(Hons), MBBS(Hons), MRCSEda; Molyneux, Samuel G. MSc, FRCSEd(Tr&Orth)a; White, Timothy O. MD, FRCSEd(Tr&Orth)a; Clement, Nicholas D. FRCSEd(Tr&Orth), PhDa; Duckworth, Andrew D. MSc, FRCSEd(Tr&Orth), PhDb; Keating, John F. MBBCh(Hons), MPhil, FRCSEd(Tr&Orth)a. Open Reduction and Internal Fixation for Humeral Shaft Nonunion: Bone Grafting Is Not Routinely Required and Avoids Donor Site Morbidity. Journal of Orthopaedic Trauma 35(8):p 414-423, August 2021. | DOI: 10.1097/BOT.0000000000002032
16. Rollo G, Prkic A, Bisaccia M, Eygendaal D, Pichierri P, Marsilio A, Giaracuni M, Meccariello L. Grafting and fixation after aseptic non-union of the humeral shaft: A case series. J Clin Orthop Trauma. 2020 Feb;11(Suppl 1):S51-S55. doi: 10.1016/j.jcot.2019.08.020. Epub 2019 Aug 31. PMID: 31992917; PMCID: PMC6977174.
17. Kumar MN, Ravindranath VP, Ravishankar M. Outcome of locking compression plates in humeral shaft nonunions. Indian J Orthop. 2013 Mar;47(2):150-5. doi: 10.4103/0019-5413.108899. PMID: 23682176; PMCID: PMC3654464.
18. Attias N, Lehman RE, Bodell LS, Lindsey RW. Surgical management of a long segmental defect of the humerus using a cylindrical titanium mesh cage and plates: a case report. J Orthop Trauma. 2005 Mar;19(3):211-6. doi: 10.1097/00005131-200503000-00011. PMID: 15758677. 11
19. Çobano?lu M, Özbey Ö, ?avk Ö, Özgezmez F. What is the Effect of 90-Degree Double-Plate Fixation with Grafting on Healing of Humeral Shaft Non-unions. Meandros Med Dent J 2016;17:83-89.
20. Hornicek FJ, Zych GA, Hutson JJ, Malinin TI. Salvage of humeral nonunions with onlay bone plate allograft augmentation. Clin Orthop Relat Res 2001; 386: 203-9.
21. Rubel IF, Kloen P, Campbell D, Schwartz M, Liew A, Myers E, et al. Open reduction and internal fixation of humeral nonunions: a biomechanical and clinical study. J Bone Joint Surg Am 2002; 84: 1315-22.
22. Oryan A, Alidadi S, Moshiri A, Bigham-Sadegh A (2014) Bone morphogenetic proteins: a powerful osteoinductive compound with non-negligible side effects and limitations. Biofactors 40(5):459–481
23. Tressler MA, Richards JE, Sofianos D, Comrie FK, Kregor PJ, Obremskey WT (2011) Bone morphogenetic protein-2 compared to autologous iliac crest bone graft in the treatment of long bone nonunion. Orthopedics 34(12):e877–e88.