Rare Case Report on Multifocal Extra Nodal Rosai-Dorfman Disease in a Treated Patient of Medulloblastoma.
Rare Case Report on Multifocal Extra Nodal Rosai-Dorfman Disease in a Treated Patient of Medulloblastoma.
Dr. Narendra Singh Butola1*, Dr Pramod S Chinder2, Dr Yogarakshith A.R3
1,3. Fellow in musculoskeletal oncology, HCG Cancer Centre, Bengaluru.
2. Head of Dept. Musculoskeletal Oncolgy, HCG Cancer Centre, Bengaluru.
*Correspondence to: Dr. Narendra Singh Butola, Fellow in musculoskeletal oncology, HCG Cancer Centre, Bengaluru. narendrabuola00@gmail.com
Copyright.
© 2024 Dr. Narendra Singh Butola. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 11 January 2024
Published: 27 January 2024
DOI: https://doi.org/10.5281/zenodo.14185278
ABSTRACT
Rationale- Rosai-Dorfman disease (RDD) is a rare histiocytic disorder typically presenting as painless cervical lymphadenopathy. Extranodal involvement is common and may also affect bones. Here we present an old treated patient of medulloblastoma, now presented with multifocal bone manifestations involving vertebral body, neck of left femur and right ischium with risk of impending fracture in the region of neck of left femur. Multifocal bone involvement is exceedingly rare and heterogeneous. Clinical outcome in terms of mortality seems to be favorable in most cases. Currently, therapy strategies include surgical and immunosuppressive treatments, but the optimal treatment of osseous RDD remains to be defined.
Patient concerns- A 31 year old female presented with complaints of generalized bony pains with pain in left hip region.
Intervention- Cemented modular bipolar hemiarthroplasty for left hip.
Lessons- This case suggests that RDD can present with multiple skeletal lesions with risk of impending fracture which needs early prophylactic intervention.
Abbreviation- RDD = Rosai-Dorfman disease.
Rare Case Report on Multifocal Extra Nodal Rosai-Dorfman Disease in a Treated Patient of Medulloblastoma.
Introduction
RDD is a rare ailment with an unknown pathophysiology that is characterised by the growth of giant histiocytes with distinct intact intracytoplasmic leukocytes (emperipolesis) and a variable mixed inflammatory infiltrate (1). Rosai-Dorfman disease (RDD), also known as sinus histiocytosis with massive lymphadenopathy, is a rare lymphocytic condition that usually manifests as painless cervical lymphadenopathy in teenagers and young adults (2). Although RDD may develop primarily in a number of extranodal sites, including skin, subcutaneous tissue, deep soft tissue, bone, head and neck, visceral organs, central nervous system, breast, thyroid, salivary glands, mesentery, and retroperitoneum, it typically manifests in lymph nodes, which are involved in most patients. 43% of cases of RDD are extranodal (3). Fever, leukocytosis, anaemia, weight loss, and polyclonal hypergammaglobulinemia are a few examples of systemic symptoms that might occur in patients of RDD (4). In less than 10% of patients with RDD, there is bone involvement. Because osseous RDD is so uncommon and has a strong inflammatory component, diagnosing it can be challenging because it is frequently mistaken for other lesions like lymphoma, Langerhans cell histiocytosis (LCH), osteomyelitis, storage disorders, and sarcoma. It may be difficult to diagnose extranodal forms or situations where there is considerable fibrosis or little emperipolesis (5). RDD has recently been recommended as a separate category ("R group") in a revised classification of histiocytic disorders and neoplasms of the macrophage-dendritic cell lineage due to its distinct features. RDD histiocytes can be distinguished from Langerhans histiocytosis by their immunohistochemical staining, which is positive for CD68, CD14, HLA-DR, fascin, CD163, and S100 and typically negative for CD1a (6).
Case Report
31 year old female presented to us with complaints of limp while walking from past few months. No history of fever, loss of appetite, weight loss or any other constitutional symptoms. Patient is a post operated case of Medulloblastoma brain and occipital craniotomy with VP shunting was done in May 2018 in London for the same along with adjuvant chemo-radiation. Patient received craniospinal radiotherapy (35Gy/21fractions) + (20Gy/12 fractions) for approx. 6 weeks in Aug-Sep 2018. Patient received 6 cycles of Cisplatin, Vincristine and Lomustine, during Sep 2018-Dec 2018. Patient was on regular follow up and regular MRI Brain was done during follow up and patient was asymptomatic during the time period of last 4 years, until last month when she developed sudden onset right lower limb limp while walking. Patient was evaluated for above mentioned complaints and relevant scans were done. On X ray (Fig. 1), MRI (Fig. 2) and PET scan (Fig. 3) multiple lytic lesion were seen involving D7 vertebra, right ischium and left proximal femur. Left proximal femur lesion was measuring 5.5 x 3.8 x 3.2 cm with a cortical breach. CT guided biopsy from D7 vertebra was done which showed typical emperipolesis (Fig. 4) S/O Rosai Dorfman Disease. IHC was done and was positive for S100 and CD68 suggestive and negative for CD1a suggestive of Rosai Dorfman Disease. High risk of pathological fracture of left proximal femur (Mirel’s Score – 11) was explained to the patient and cemented bipolar hemiarthroplasty was done for left hip. Post op patient was mobilized on Day 1 and dressing was changed at Day 5 and patient was discharged at Day 6. Final histopathology impression was s/o Extranodal Rosai Dorfman disease.
Fig. 1(a) Pre op X ray (b) Post op X ray
Fig. 2 (a) MRI Coronal section (b) Axial Section
Fig. 4 Histopathological images showing emperipolesis in RDD
Discussion
Massive bilateral painless cervical lymphadenopathy with a fever, weight loss, and night sweats are the hallmark symptoms of classic RDD (7). Infectious factors, such as the herpes virus, parvovirus B19, Epstein Barr virus, or even bacterial infections, immunodeficiency or autoimmune illnesses, are being considered as potential causes of RDD (idiopathic disease), however the specific cause is yet unknown (8). Skin (10%), nasal cavity (11%), bone (5%–10%), orbital tissue (11%) and the central nervous system (5%), among other extra-nodal locations, are frequently affected. The cranial and facial bones, as well as the long bones, are the most commonly affected areas in osseous lesions (9). Due to the heterogeneous chronic inflammatory infiltration and associated fibrosis and focal osteonecrosis, RDD was most frequently mistaken for granulomatous disease or osteomyelitis. The diagnosis cannot be guided by radiological imaging such as a CT scan or an MRI because they lack specificity (10). RDD diagnosis is never an easy task. Because our patient's medulloblastoma had already been detected and treated with chemotherapy and radiation, our case was more difficult. The diagnosis must be made through histological analysis since emperipolesis is a defining but variable feature. Immunohistochemical staining is also crucial because RDD is characterised by the expression of S-100. A fraction of patients with RDD has been found to contain BRAF, NRAS, KRAS, MAP2K1, or ARAF mutations, according to recent investigations. This could be interpreted as supporting the idea that RDD is a heterogeneous entity with both immunological and malignant manifestations. Treatment guidelines for RDD are still not delineated and patients are managed symptomatically or according to clinical manifestation (11). 20 to 50 % cases of RDD with asymptomatic cutaneous manifestation can be managed with observation alone and show spontaneous remission (12). Surgical excision is effective in unifocal disease. Surgical resection in multifocal disease is only advisable for the lesions with neurologic or end organ dysfunction. Role of steroids, chemotherapy, immunotherapy and radiation in RDD is still under research with variable efficacy depending upon the subtype (11). Prognosis in RDD is variable, cutaneous RDD usually shows favorable prognosis whereas prognosis in RDD with multifocal lesions or multisystem involvement is usually poor.
This case is worth notifying because of two reasons first there is a prior history of chemoradiation for a brain medulloblastoma and as there is no literature review accessible in this area, further discussion regarding the relationship between medulloblastoma and RDD is needed, second reason is multifocal nature of our case. In our case skeletal lesions are seen involving femur, pelvis and vertebrae which is very rarely seen in RDD.
Conclusion
RDD is a rare disorder that can present as multiple skeletal lesions in patients with a history of malignancy. The diagnosis is made based on histopathology and immunohistochemistry, and treatment depends on the extent and severity of the disease. Our case highlights the importance of considering RDD in the differential diagnosis of patients with multiple skeletal lesions and a history of malignancy.
References
1.Adam R, Harsovescu T, Tudorache S, Moldovan C, Pogarasteanu M, Dumitru A, Orban C. Primary Bone Lesions in Rosai-Dorfman Disease, a Rare Case and Diagnostic Challenge-Case Report and Literature Review. Diagnostics (Basel). 2022 Mar 23;12(4):783. doi: 10.3390/diagnostics12040783. PMID: 35453831; PMCID: PMC9032234.
2.Jiang H, Song J, Lin W, Yi M, Yao M, Ding L. Rosai-Dorfman disease with spine involvement: A case report. Medicine (Baltimore). 2022 Feb 25;101(8):e28413. doi: 10.1097/MD.0000000000028413. PMID: 35212270; PMCID: PMC8878865.
3.Sassi F, M'rad H, Kacem LB, Sassi B, Hannachi S, Rammeh S. A lesion of the patella: An unexpected location of Rosai-Dorfman disease: A case report. Int J Surg Case Rep. 2022 Sep;98:107510. doi: 10.1016/j.ijscr.2022.107510. Epub 2022 Aug 13. PMID: 35985119; PMCID: PMC9411659.
4.Garcia RA, DiCarlo EF. Rosai-Dorfman Disease of Bone and Soft Tissue. Arch Pathol Lab Med. 2022 Jan 1;146(1):40-46. doi: 10.5858/arpa.2021-0116-RA. PMID: 34965285.
5.Demicco EG, Rosenberg AE, Björnsson J, Rybak LD, Unni KK, Nielsen GP. Primary Rosai-Dorfman disease of bone: a clinicopathologic study of 15 cases. Am J Surg Pathol. 2010 Sep;34(9):1324-33. doi: 10.1097/PAS.0b013e3181ea50b2. PMID: 20679880.
6.Mosheimer BA, Oppl B, Zandieh S, Fillitz M, Keil F, Klaushofer K, Weiss G, Zwerina J. Bone Involvement in Rosai-Dorfman Disease (RDD): a Case Report and Systematic Literature Review. Curr Rheumatol Rep. 2017 May;19(5):29. doi: 10.1007/s11926-017-0656-6. PMID: 28401384; PMCID: PMC5388731.
7.Bruce-Brand C, Schneider JW, Schubert P. Rosai-Dorfman disease: an overview. J Clin Pathol. 2020 Nov;73(11):697-705. doi: 10.1136/jclinpath-2020-206733. Epub 2020 Jun 26. PMID: 32591351.
8.Delacre taz F, Meuge -Moraw C, Anwar D, Borisch B, Chave JP. Sinus histiocytosis with massive lymphadenopathy (Rosai Dorfman disease) in an HIV-positive patient. Virchows Arch A Pathol Anat Histopathol. 1991;419(3) : 251-254.
9.Cohen Aubart F, Idbaih A, Emile JF, Amoura Z, Abdel-Wahab O, Durham BH, Haroche J, Diamond EL. Histiocytosis and the nervous system: from diagnosis to targeted therapies. Neuro Oncol. 2021 Sep 1;23(9):1433-1446. doi: 10.1093/neuonc/noab107. PMID: 33993305; PMCID: PMC8408883.
10.Patel MH, Jambhekar KR, Pandey T, Ram R. A rare case of extra nodal Rosai-Dorfman disease with isolated multifocal osseous manifestation. Indian J Radiol Imaging. 2015 Jul-Sep;25(3):284-7. doi: 10.4103/0971-3026.161459. PMID: 26288524; PMCID: PMC4531454.
11.Abla O, Jacobsen E, Picarsic J, Krenova Z, Jaffe R, Emile JF, Durham BH, Braier J, Charlotte F, Donadieu J, Cohen-Aubart F, Rodriguez-Galindo C, Allen C, Whitlock JA, Weitzman S, McClain KL, Haroche J, Diamond EL. Consensus recommendations for the diagnosis and clinical management of Rosai-Dorfman-Destombes disease. Blood. 2018 Jun 28;131(26):2877-2890. doi: 10.1182/blood-2018-03-839753. Epub 2018 May 2. PMID: 29720485; PMCID: PMC6024636.
12.Pulsoni A, Anghel G, Falcucci P, et al. Treatment of sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease) : report of a case and literature review. Am J Hematol. 2002;69(1):67-71
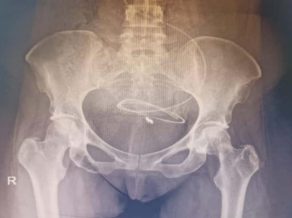
Figure 1
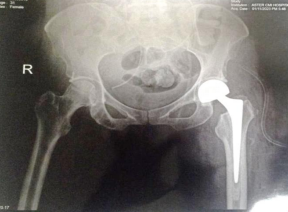
Figure 2
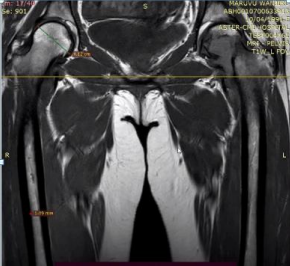
Figure 3
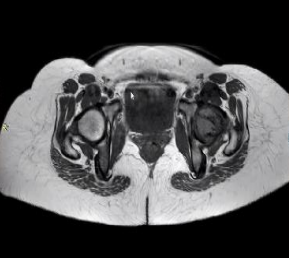
Figure 4
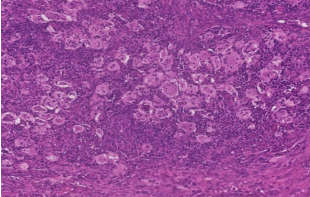
Figure 5
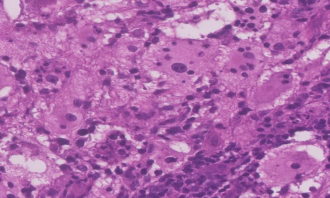
Figure 6
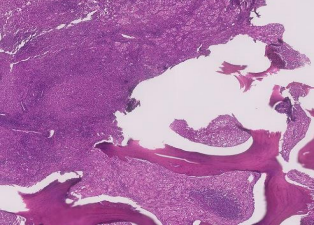
Figure 7
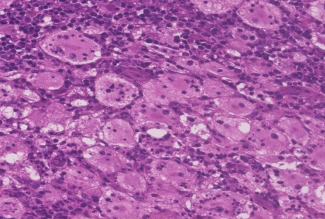
Figure 8