One Month Old Girl with Extensive Diffusse Congenital Hemangioma.
One Month Old Girl with Extensive Diffusse Congenital Hemangioma.
Dr. Ahmed Moosa Yahya Assery *
*Correspondence to: Dr. Ahmed Moosa Yahya Assery, Paediatrics ambulatory care consultant. King Abdulaziz Medical City, National Guard Medical Affairs, Family Medicine-Ambulatory care, NGCSC.
Copyright
© 2024: Dr. Ahmed Moosa Yahya Assery. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 06 November 2015
Published: 15 January 2016
Abstract
Congenital haemangiomas are one of the confusing skin manifestations not only due to the cosmetic reasons but also since of the associated neurological medical problems that comes with one of them is sturge-weber syndrome that presents with early seizures. Congenital haemangiomas are vascular lesions that are fully formed at birth and occur when blood vessels form abnormally. The cells that form blood vessels are called endothelial cells. In a congenital haemangioma, these cells multiply more than they should. The extra tissue forms a benign tumour attached to normal blood vessels. The cause is unknown. (1). Our interesting case as you will see shows a extremely diffuse congenital haemangioma that is so striking and impressive till a degree the newborn admitted in the hospital for further investigations one of them brain magnetic resonance imaging. Here in this article, I will high light about some aspects of the huge congenital haemangiomas published and their associated sequalae.
Keywords: Haemangioma: HMG.
Congenital Haemangioma: CHMG.
Rapidly involuting congenital Haemangioma: RICH.
Non- involuting congenital haemangioma: NICH.
partially involuting congenital haemangioma: PICH.
Sturge-Weber syndrome: SWS.
Port-Wine Stain: PWS.
Magnetic Resonance Imaging: MRI.
Guanin nucleotide-binding protein: GNA.
One Month Old Girl with Extensive Diffusse Congenital Hemangioma.
Introduction
Congenital haemangiomas (CHMGs) are rare, benign vascular tumours that are present and fully grown at birth. They present as bossed plaques or exophytic masses located on the head, neck, or limbs. Based on their natural history, three major clinical subtypes of CH have been recognized: rapidly involuting congenital haemangioma (RICH); non- involuting congenital haemangioma (NICH); and partially involuting congenital haemangioma (PICH), which shows overlapping features and clinical behaviour of RICH and NICH.
There is increasing evidence that most, if not all, CHMGs, whether RICH, or PICH, are due to somatic activating mutations in GNAQ and its paralog GNA11. This suggests that RICH, NICH, and PICH represent a disease spectrum rather than separate entities.
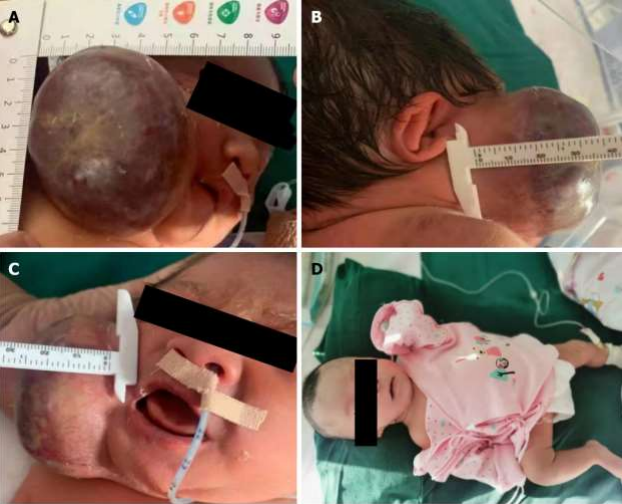
Clinically CHMGs are present and fully grown at birth. They usually present as solitary, plaque-like or exophytic lesions varying from a few centimetres to >10 cm in size (picture 1A-D).
Based on their natural history, three major clinical subtypes of CH have been recognized:
-RICH.
- NICH.
- PICH.
In most cases, RICH start rapidly regressing in the few weeks after birth and involute completely or nearly completely by the age of 14 months. NICH do not involute but typically grow in proportion with somatic growth. PICH show clinical and behavioural features of both RICH and NICH; they undergo a phase of rapid involution and then stabilize without regressing completely. Several reports have added another dimension to the natural history of CHs: those that continue to expand and progress over time.
The differential diagnosis of congenital CHMG includes other benign vascular tumours, vascular malformations, and benign and malignant nonvascular tumours as follow:
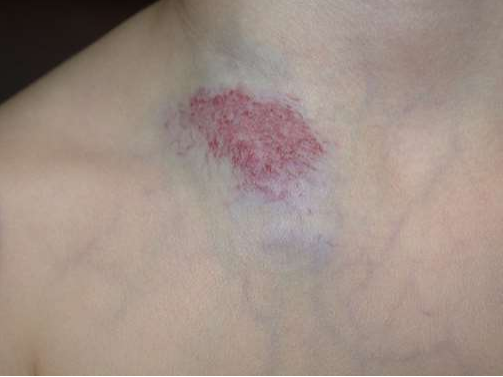
- Infantile haemangioma: a flat area of discoloration was present at birth, since the latter could represent a premonitory mark of infantile haemangioma (picture 2).
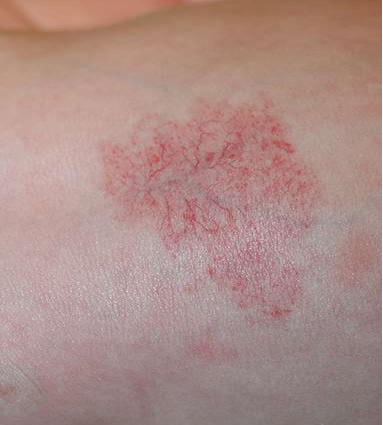
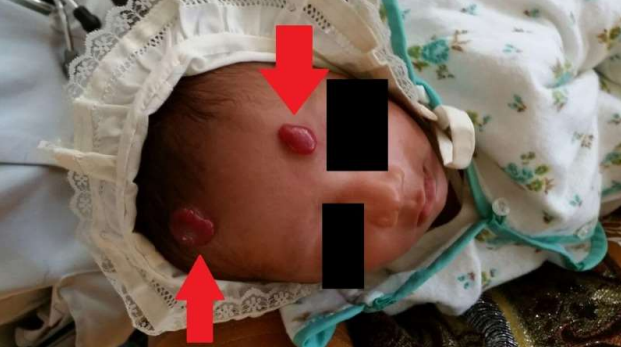
- Tufted angioma – Tufted angioma is an uncommon, benign vascular tumour that usually develops in early infancy but may be present at birth. It presents as an infiltrated, firm, dusky, red to violaceous plaque or nodule with typical, overlying hypertrichosis (picture 3).
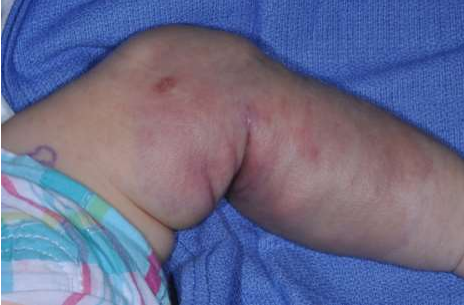
- Kaposiform hemangioendothelioma – Kaposiform hemangioendothelioma is a rare vascular tumour that can be present at birth. Kaposiform hemangioendothelioma appears as a slightly raised, subcutaneous, firm mass with a purpuric, bruised appearance (picture 4).
- Infantile myofibromatosis/hemangiopericytoma:
- Lipoblastoma and lipoblastomatosis.
- Malignant tumours. (2).
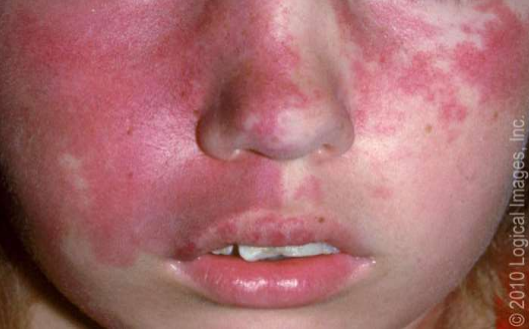
- SWS: In this article being taking about CHMG we must pass by the SWS, so what is this syndrome? It is a rare congenital vascular disorder characterized by facial capillary malformation (port wine birthmark) and associated capillary-venous malformations affecting the brain and eye. It is not a heritable disorder. Thus, recurrence in another relative in the same or future generations is unlikely. The main cause of SWS is somatic mosaic GNAQ pathogenic variants, as identified in a study that performed whole-genome sequencing of affected and normal tissue samples from three patients with SWS. Clinically SWS is characterized by a facial capillary malformation, also known as a port wine birthmark (Picture 5), and an associated leptomeningeal capillary-venous malformation (leptomeningeal angioma) involving the brain and eye. These vascular malformations are associated with specific neurologic and ocular abnormalities.
The neurologic features of SWS may be progressive and include seizures, focal neurologic deficits, and intellectual disability. Visual field defects are common when the occipital cortex is affected. These manifestations occur with variable severity. Hydrocephalus also may occur. This complication is thought to result from increased venous pressure caused by thrombosis of the deep venous channels or extensive arteriovenous anastomoses. (3).
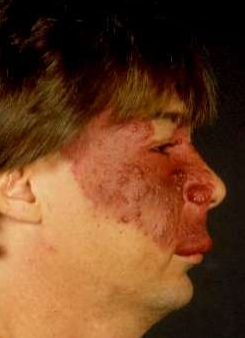
- PWS — Cutaneous port wine stain (capillary malformation) is the most common type of vascular malformation, occurring in 0.3 percent of newborn infants (picture 6). However, only a small proportion of children with port wine birthmarks have SWS.
In SWS, the port wine birthmark typically is present on the forehead and upper eyelid, primarily in the distribution of the first or second division of the trigeminal nerve. In one report, 274 patients had port wine birthmarks located in the distribution of the trigeminal nerve, and involvement of the eye or central nervous system was noted in 22 (8 percent). All 22 patients of these patients had a port wine birthmark in the first or second division of the trigeminal nerve. Although overlapping the trigeminal distribution, the pattern of capillary malformations in SWS may reflect the embryonic vascular supply of the face. Although traditionally considered important in determining meningeal involvement risk, the location of the capillary malformation (port wine birthmark) on the face is not a good indicator of the risk of an underlying meningeal capillary malformation. Extension of the skin lesion to both sides of the face and the trunk and extremities is common.
The skin lesion usually is obvious at birth. However, its appearance changes with age and its size increases as the patient grows. In the newborn, the lesion is flat and usually light pink in colour. It typically darkens with age to a deep red, port wine appearance, and vascular ectasias may develop (picture 6). The vascular ectasias produce nodularity and superficial blebbing, which lead to overgrowth of the underlying soft tissues and sometimes the bone. Dental abnormalities may occur because of gingival thickening if this area is affected. By the fifth decade, 65 percent of patients with a facial port wine birthmark have hypertrophy and nodularity within the lesion.
(PICTURE 1-A: Rapidly involuting congenital Haemangioma).
(PICTURE 1-B: Rapidly involuting congenital Haemangioma).
(PICTURE 1-C: Non- involuting congenital haemangioma).
(PICTURE 1-D: Non- involuting congenital haemangioma).
(PICTURE 2: Infantile haemangioma).
(PICTURE 3: Tufted angioma).
(PICTURE 4: Kaposiform hemangioendothelioma)
(PICTURE 5: Sturge Weber Syndrome).
(PICTURE 6: Port Wine Stain).
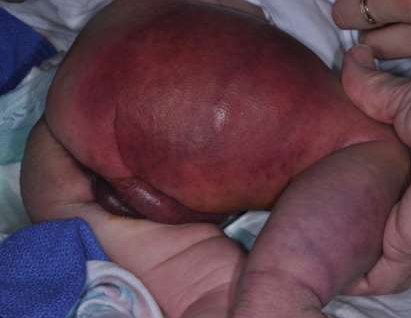
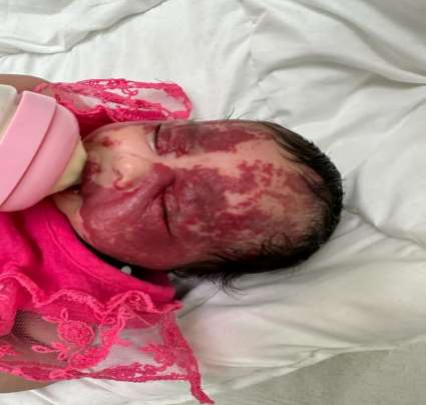
(PICTURE 7: Our current case presentation and obvious here the massive reddish PWS on the face)
(PICTURE 8: 3 weeks old girl with congenital haemangioma).
(PICTURE 9: A pre-term baby with giant congenital haemangioma).
Please click here to view all pictures
Our Case Presentation
This newborn who is the product of first degree consanguinity and totally negative family history of any skin lesions (specially HMGs) has delivered as a full term baby with spontaneous vaginal delivery and regular prenatal maternal follow up with prenatal obstetric clinic that was unremarkable for anything on the fetus and the mother, the newborn appearance on the delivery room was so impressive for the medical team since of the giant, extensive and diffuse reddish lesion all over the body (Picture 7).no peri-natal events recorded like seizures or respiratory distress. The child kept in the word for further investigations and consultations as per recommended by the dermatology team who labelled the diagnose initially as PWS to role out SWS.
The booked brain of the child showed a normal findings and absence of any abnormal vascular lesions in the brain and the spinal cord. The neurology, cardiology and ophthalmology teams reported any abnormal finding that could be complicated by this massive skin lesion. The patient after which discharged home with further follow up with dermatology team.
Discussion about the Case:
As you saw previously despite the massive PWS of our case but fortunately no associated SWS elements found as we were suspecting initially and as mentioned in many references (one of them is the trustable Up to Date (3)), that mentioned about the so weak associations between the PWS and SWS. though of that PWS represents the most common type of vascular malformation, occurring in 0.3 percent of newborn infants.
Generally, the precise incidence of CHMG is unknown. In a prospective study, RICH occurred in 2 of 594 newborns (0.3 percent). In a retrospective review of 6459 children with vascular anomalies seen in a vascular anomalies centre, CHMGs were diagnosed in 14 percent, infantile haemangiomas were diagnosed in 43 percent, and capillary malformations were diagnosed in 30 percent. There are no known risk factors for CHMG. In contrast with infantile haemangioma, there is no association between CHMG and premature birth or multiple gestation (2).
In (4), Int J Mol Sci. 2019 May; 20(9): 2243. Published online 2019 May 7. doi: 10.3390/ijms20092243 published by Vi Nguyen,1 Marcelo Hochman,2 Martin C. Mihm, Jr.,3 J. Stuart Nelson,4,5 and Wenbin Tan on 2019 what was mentioned in that association is : Approximately 15–20% of children a facial PWS involving the ophthalmic trigeminal dermatome are at risk for SWS a neurocutaneous disorder with vascular malformations in the cerebral cortex on the same side of the facial PWS lesions. The prevalence of PWS is estimated at three to five children per 1000 live births; there are ~1.2 million individuals in the United States and ~26 million people worldwide with PWS birthmarks. There is no sex predilection, and the inheritance pattern is generally sporadic. Approximately 90% of PWS are located on the face, followed by neck, trunk, and extremities at much less frequencies. The majority of facial PWS (~90%) are unilateral in a trigeminal dermatomal distribution.
Among the search about the similar case reports of CHMGs I found such case reports:
The first one published in pumped ventral journal Cureus. 2018 Apr; 10(4): e2485.Published online 2018 Apr 16. doi: 10.7759/cureus.2485 by Kamleshun Ramphul, 1 Stephanie G Mejias,2 Yogeshwaree Ramphul-Sicharam,3 and Ruhi Sonaye4 about this infant (Picture 8)(5) who was 3 weeks old with CHMG and the article concluded that CHMGs have a low incidence rate, and it is vital for every physician to properly diagnose this condition. While usually, most physicians prefer to adopt a wait-and-watch approach, a proper differential diagnosis to rule out other conditions with similar symptoms should be appropriately performed. For CHMGs that do not regress, surgical excision can be considered once the child is of pre-school age.
1 Stephanie G Mejias,2 Yogeshwaree Ramphul-Sicharam,3 and Ruhi Sonaye4 about this infant (Picture 8)(5) who was 3 weeks old with CHMG and the article concluded that CHMGs have a low incidence rate, and it is vital for every physician to properly diagnose this condition. While usually, most physicians prefer to adopt a wait-and-watch approach, a proper differential diagnosis to rule out other conditions with similar symptoms should be appropriately performed. For CHMGs that do not regress, surgical excision can be considered once the child is of pre-school age.
The second one (6) published in world journal of clinical cases, World J Clin Cases. Jun 16, 2022; 10(17): 5756-5763 Published online Jun 16, 2022. doi: 10.12998/wjcc. v10.i17.5756, Ren N, Jin CS, Zhao XQ, Gao WH, Gao YX, Wang Y, Zhang YF. Preterm neonate with a large CHMG on maxillofacial site causing thrombocytopenia and heart failure: A case report. World J Clin Cases 2022; 10(17): 5756-5763 [PMID: 35979094 DOI: 10.12998/wjcc.v10.i17.5756] by Baishideng Publishing Group Inc. under the title Preterm neonate with a large CHMG on maxillofacial site causing thrombocytopenia and heart failure: A case report (Picture 9) that said CHMGs are significantly different from typical haemangiomas in terms of the clinical manifestations, staging, pathology, and imaging findings. CHMGs are of different types, NICH, RICH, and PICH. The treatment strategies, incidence of complications, and long-term prognosis are also different. Therefore, it is crucial to determine the type of CHMG based on the clinical characteristics, colour Doppler ultrasonography, and imaging. The treatment strategy should be guided by the specific type. Common complications of CHMG include intralesional haemorrhage, thrombocytopenia, abnormal coagulation function, and congestive heart failure. In our patient, we focused on limiting the liquid intake, inhibiting further growth of the haemangioma, alleviating the congestive heart failure, improving heart function, supplementing Hb, preventing bleeding, and selecting the timing for the surgery. Furthermore, close multidisciplinary collaboration, meticulous care of the tumour, surgical planning, and postoperative care were instrumental in averting postoperative complications.
The third one (7) published on ultrasound in obstetrics and gynaecology. Volume40, IssueS1 07 September 2012 by I. Martinez-Wallin, R. Rayse, P. Anabel, T. Juan Mario under the title Congenital venous hemangioma: a case report that admitted the benefit of using 3 and 4 dimensions sonographies to diagnose and some times treating the diagnosed pre-natal CHMGs.
Conclusion
It is a fact that despite the scarcity of the CHMGs particularly the massive ones including the PWS ones. Still the associated SWS is so rare, but that does not mean the importance of the work up of any coming CHMGs from the neurological, ophthalmological and cardiological sides.
References
1_ -https://www.hopkinsmedicine.org/health/conditions-and-diseases/congenital-hemangioma.
2- https://www.uptodate.com/contents/congenital-hemangiomas-rapidly-involuting-congenital-hemangioma-rich-noninvoluting-congenital-hemangioma-nich-and-partially-involuting-congenital-hemangioma-pich?search=congenital%20hemangioma&source=search_result&selectedTitle=1%7E150&usage_type=default&display_rank=1.
3-https://www.uptodate.com/contents/sturge-weber-syndrome?search=sturge%20weber&source=search_result&selectedTitle=1%7E34&usage_type=default&display_rank=1.
4-https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6539103/.
5- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6003795/.
6-https://www.wjgnet.com/2307-8960/full/v10/i17/5756.htm.
7- https://doi.org/10.1002/uog.12087.