Clinical Image Interest of Corneal OCT in Lens Dislocation
Clinical Image Interest of Corneal OCT in Lens Dislocation
Lucrèce Joanelle Vydalie ERIGA1* , Keith Adjatin Rolyf2 , Arnaud Hugues Yempabou YONLI3, Fadhloullahi Khidrou Sambou OUMAROU4, Yassine MOUZARI5, Abdelbarre OUBAAZ6.
*Correspondence to: Lucrèce Joanelle Vydalie ERIGA. Department of ophthalmology,Hôpital Militaire d’instruction Mohammed V-Rabat, Morroco.
Copyright
© 2024 Lucrèce Joanelle Vydalie ERIGA. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 03 May 2024
Published: 20 May 2024
Clinical Image Interest of Corneal OCT in Lens Dislocation
Clinical Cases
This was a patient aged around 60 with no particular history, who consulted the emergency department for a painful red eye associated with a drop in visual acuity, all occurring in the context of a domestic trauma (inadvertent elbow strike) that had occurred around 8 hours earlier.
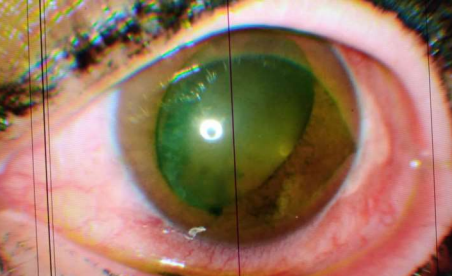
Examination of the right eye revealed visual acuity at 30 centimetres, good palpebral statistics, diffuse conjunctival hyperhemia, a clear cornea, and a negative fluorescein test. The anterior chamber was inferiorly narrow with a Van Herrick grade II. The iris had good trophicity, the pupil was distorted (oval) and the lens was partially dislocated in the anterior chamber. Intraocular pressure was 18mmhg. The fundus, within the limits of the examination, was unremarkable.
Examination of the left eye was unremarkable.
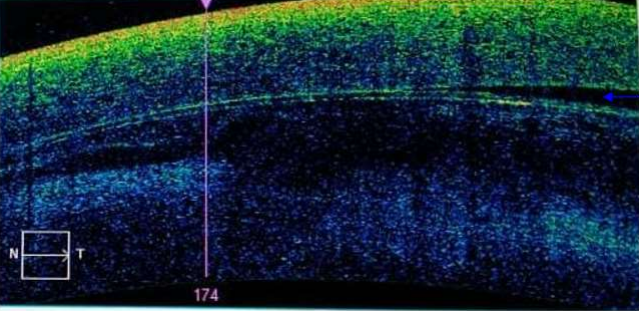
The OCT (Optical Coherence Tomography) carried out in search of complications showed contact between the lens and the endothelium. Given the results of the OCT, the patient underwent emergency surgery.
Image1: Lens subluxation, oval pupil
Image 2: OCT of the cornea showing partial contact of the lens with the corneal endothelium (blue arrow indicating absence of contact).