An Unusual Presentation of Non-Small Cell Carcinoma of Lung
An Unusual Presentation of Non-Small Cell Carcinoma of Lung
Dr. Reuben Jacob *1, Dr. Midhun2, Dr. Kesavan V Nair3
1. Dr Reuben Jacob, Second Year M.D Respiratory Medicine Resident, Department of Respiratory Medicine, Sree Uthradom Thirunal Academy of Medical Sciences, Thiruvananthapuram, Kerala, India.
2. Dr Midhun J, DNB Respiratory Medicine, Senior Resident in the Department of Respiratory Medicine, Sree Uthradom Thirunal Academy of Medical Sciences, Thiruvananthapuram, Kerala, India.
3. Dr. Kesavan V Nair, Professor, Department of Respiratory Medicine, Sree Uthradom Thirunal Academy of Medical Sciences, Thiruvananthapuram, Kerala, India.
Correspondence to: Dr Reuben Jacob, Second Year M.D Respiratory Medicine Resident, Department of Respiratory Medicine, Sree Uthradom Thirunal Academy of Medical Sciences, Thiruvananthapuram, Kerala, India.
Copyright
© 2024 Dr. Reuben Jacob. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 17 May 2024
Published: 03 June 2024
ABSTRACT
Introduction: Pulmonary adenocarcinoma is a type of lung cancer which is often misdiagnosed due to its unusual presentation mimicking both infectious and inflammatory diseases of the lung. In this case report we describe about the patient with lung adenocarcinoma who had a rapid deterioration in his respiratory status.
Case Description: 53-year-old male was referred to our centre as there was no radiological resolution after being treated for pneumonia. Chest X-ray later taken showed a classic ‘Golden S’ sign which raised a suspicion of malignancy later HRCT thorax showed a ‘bronchial cut off’ sign. Bronchoscopy biopsy confirmed Non-Small Cell Carcinoma- Adenocarcinomatous type.
Conclusion: This case stresses the importance of radiology in early diagnosis of bronchogenic carcinoma even from peripheral centres. Chest Xray still remains the key in arriving at a diagnosis in respiratory medicine.
An Unusual Presentation of Non-Small Cell Carcinoma of Lung
Introduction
Lung cancer, among the most common malignancies, is categorized into small-cell lung cancer (SCLC) and non-small-cell lung cancer (NSCLC), with the latter being more prevalent1. Pulmonary adenocarcinoma falls under the NSCLC category and is strongly linked to prior smoking. Typically located in the lung periphery, lung adenocarcinoma is often found in areas of scarring or chronic inflammation. It primarily originates in mucosal glands and constitutes approximately 40% of all lung malignancies2. The average age of diagnosis for lung adenocarcinoma is 71 years, and its occurrence before the age of 20 is uncommon.
Case Description
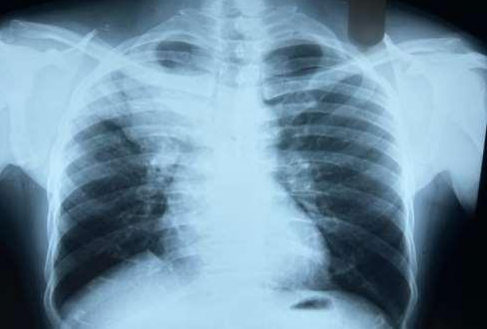
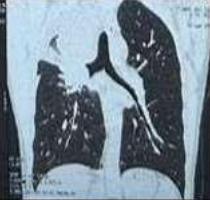
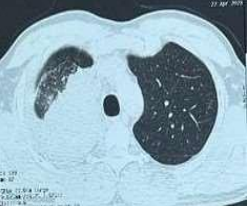
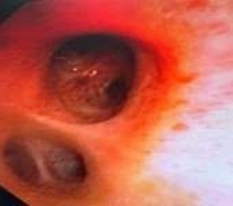
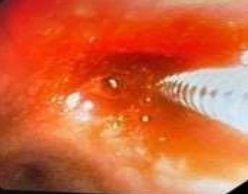
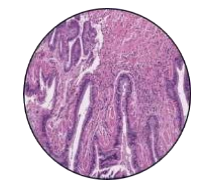
53-year-old male presented to a tertiary care centre elsewhere with complaints of high-grade fever not associated to chills/rigor, productive cough with copious amount of white coloured sputum, breathing difficulty on exertion MMRC Grade III, noisy breathing, chest pain and multiple episodes of haemoptysis. Patient had no other co-morbidities/symptoms of any chronic illness or any other contributary past history. After getting admitted and primary investigations, he was treated with IV antibiotics suspecting pneumonia. After 7 days of treatment patient was referred to our centre as there was no resolution. On clinical examination, patient had shift of trachea to the right side, dullness on percussion, bronchial breath sound and crepitations in the right infraclavicular area. Blood investigations showed leucocytosis (14,000/mm3), elevated levels of ESR (85mm/hr) and CRP (102mg/dl) with no other significant biochemical changes. Chest X-ray taken showed a collapse of the right upper lobe, with the classical ‘Golden S’ sign (figure 1) which raised a suspicion of right upper lobe mass. Further HRCT thorax was taken which showed a ‘bronchial cut off’ sign (figure 2) and an enhancing soft tissue lesion in the right upper lobe with encasement right pulmonary artery and bronchus (figure 3). Patient was taken up for bronchoscopy which showed a right upper lobe mass (figure 4). Bronchial brushings and biopsy (figure 5) were taken and samples were sent for Histopathological study which confirmed Non-Small Cell Carcinoma- Adenocarcinomatous type (figure 6).
Figure 1: Chest-Xray
Figure 2: CT image showing Bronchial cut-off sign
Figure 3: CT image showing right upper lobe mass
Figure 4: Bronchoscopy image showing right upper lobe mass
Figure 5: Bronchoscopy Biopsy
Figure 6: Histopathology
Discussion
Established risk factors for lung adenocarcinoma includes a family history of the disease, cigarette smoking, and exposure to occupational hazards. These factors contribute to genetic alterations in the p53 gene, observed in up to 52% of cases, leading to the development of non-small-cell lung cancer (NSCLC)3. Additional gene alterations, such as EGFR, KRAS, and ALK, are commonly associated with NSCLC4. Over the past four decades, there has been a notable increase in the incidence of lung adenocarcinoma among women, primarily attributed to smoking5. Approximately 2% to 5% of all lung cancer cases occur in individuals under 40 years old6. Among all lung cancer types, 85% are classified as non-small cell, comprising adenocarcinomas, squamous cell carcinomas, and giant cell undifferentiated carcinomas7. Lung adenocarcinoma may manifest as ground glass nodules, consolidative opacity, or solid mass lesions on computed tomography (CT)8. As the most prevalent type of lung cancer, adenocarcinoma is associated with tobacco use9,10. It is also frequently diagnosed in nonsmokers, especially women.
Conclusion
This case highlights the importance of radiology in early diagnosis of bronchogenic carcinoma. This simple investigation is available even in peripheral centres all over India. Though Roentgen introduced CXR way back in late 1890s, it still remains the key in arriving at a diagnosis in respiratory medicine.
Ethics Approval and Consent of Participation
Case report has been written after taking an informed consent from patient. The case report was an observational study and hence ethics committee approval has been waived off.
Consent for Participation
The authors are giving complete authorisation for the journal to publish the article under first and second author’s name in the journal.
Data and Material Availability
The data and material for the case report has been obtained from the patient with his consent.
Funding No grant or financial support taken from any outside sources.
Competing Interest No conflicts of interest.
Acknowledgements
1) Department Of Pathology, Sree Uthradom Thirunal Academy of Medical Sciences, Thiruvananthapuram, Kerala, India.
2) Department Of Radiology, Sree Uthradom Thirunal Academy of Medical Sciences, Thiruvananthapuram, Kerala, India.
References
1. Myers DJ, Wallen JM. Lung Adenocarcinoma. [Updated 2023 Jun 12]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK519578/
2. Marin AC, Prasad A, Patel V, Lwoodsky C, Hechter S, Imtiaz A, Patel P, Shah V, Appiah J, Cheriyath P. Pulmonary Adenocarcinoma Mimicking Pneumonia in a Young Adult. Cureus. 2023 Feb 21;15(2):e35267. doi: 10.7759/cureus.35267. PMID: 36968868; PMCID: PMC10035766.
3. Noninvasive pulmonary nodule characterization using transcutaneous bioconductance: preliminary results of an observational study. Gariani J, Martin SP, Hachulla AL, et al. Medicine (Baltimore) 2018;97:0. [PMC free article] [PubMed] [Google Scholar]
4. Yang S, Yu X, Fan Y, Shi X, Jin Y. Clinicopathologic characteristics and survival outcome in patients with advanced lung adenocarcinoma and KRAS mutation. J Cancer. 2018;9(16):2930-2937. [PMC free article] [PubMed]
5. High incidence of EGFR mutations in pneumonic-type non-small cell lung cancer. Liu J, Shen J, Yang C, He P, Guan Y, Liang W, He J. Medicine (Baltimore) 2015;94:0. [PMC free article] [PubMed] [Google Scholar]
6. Lung cancer in patients under the age of 40 years. Kozielski J, Kaczmarczyk G, Por?bska I, Szmygin-Milanowska K, Go?ecki M. Contemp Oncol (Pozn) 2012;16:413–415. [PMC free article] [PubMed] [Google Scholar]
7. Classification and pathology of lung cancer. Zheng M. Surg Oncol Clin N Am. 2016;25:447–468. [PubMed] [Google Scholar]
8. Stark P. High Resolution Computed Tomography of the Lungs. UptoDate. 2022. https://www.uptodate.com/contents/high-resolution-computed-tomography-of-the-lungs#!
9. Paliwal P, Rajappa S, Santa A, Mohan M, Murthy S, Lavanya N. Clinical profile and outcomes of patients with Stage IV adenocarcinoma of lung: A tertiary cancer center experience. Indian J Cancer. 2017 Jan-Mar;54(1):197-202. [PubMed]
10. Rami-Porta R, Call S, Dooms C, Obiols C, Sánchez M, Travis WD, Vollmer I. Lung cancer staging: a concise update. Eur Respir J. 2018 May;51(5) [PubMed].