A Rare Case Report on Cervical Carcinosarcoma, Clinical Presentation and Treatment Outcome
A Rare Case Report on Cervical Carcinosarcoma, Clinical Presentation and Treatment Outcome
Dr. Kanika Gupta. MBBS, MS 1, Dr. Neera Agrawal 2, Dr Sanjeev Arora - MBBS, MS 3, Dr Aayushi Ruia – MBBS , MS *4
1.Senior Director, Department of Gynae – oncology, Max Super Speciality Hospital, Vaishali and Patparganj, Uttar Pradesh, India.
2.HOD, Obstetrics and Gynaecology Max Hospital, Patparganj, New Delhi.
3.Senior Consultant, Surgical Oncology Max Super Speciality Hospital, Vaishali and Patparganj, Uttar Pradesh, India.
4.Fellow in Gynae Oncology Max Super Speciality Hospital, Vaishali and Patparganj, Uttar Pradesh, India.
*Correspondence to: Dr Aayushi Ruia. Fellow in Gynae Oncology Max Super Speciality Hospital, Vaishali and Patparganj, Uttar Pradesh, India
Copyright
© 2024 Dr Aayushi Ruia. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 27 August 2024
Published: 20 September 2024
DOI: https://doi.org/10.5281/zenodo.13821560
Abstract
Cervical carcinosarcoma (CCS) also known as malignant mixed mesodermal tumor (MMMT) is a rare aggressive biphasic tumor which has high grade epithelial and mesenchymal components. Carcinosarcomas occur most frequently in uterine corpus, can also arise in ovaries, cervix, fallopian tubes, vagina, peritoneum, and extra genital sites. The Cancer Genome Atlas (TCGA) data supports conversion and combination theories where carcinomatous cells convert themselves in sarcomatous via epithelial to mesenchymal transition. Currently there has been no consensus regarding survival, management and prognosis of CCS. Here we report a case of a perimenopausal female with abnormal uterine who was diagnosed preoperatively on biopsy as poorly differentiated carcinoma, was taken for radical hysterectomy and was diagnosed on final histopathology as carcinosarcoma of cervix.
A Rare Case Report on Cervical Carcinosarcoma, Clinical Presentation and Treatment Outcome
Introduction
CCS commonly occurs in the postmenopausal women. Compared to cervical squamous cell carcinoma and adenocarcinoma, primary CCS has a worse outcome because it is prone to metastasis and recurrence. CCS is commonly confused with uterine carcinosarcoma and is easily misdiagnosed with other histologies like cervical cancers/sarcomas. However, its histological presentations, biologic behavior and outcome is different from these malignancies. Until now, the management for CCS has been mainly extrapolated from studies of uterine carcinosarcoma or cervical sarcomas, but their management remains a challenge.
Case Report
A 50-year-old perimenopausal woman presented to us in office with chief complaints of heavy bleeding during menses and had progesterone releasing intrauterine device in situ since last 4 years. She had taken medical management in the past year but was not responsive. She was multiparous, had no medical comorbidities and was a nonalcoholic, nonsmoker. MRI was suggestive of heterogenous enhancing endocervical mass of around 4.7 x 4.5 x 2.3 cm which was extending up till lower uterine segment and on clinical examination cervix was barrel shaped with bilateral parametrium and rectal mucosa free so her clinical staging was IB. We took her cervical and endometrial biopsy which was reported as poorly differentiated cervical carcinoma. Endometrium demonstrated hyperplastic changes. After evaluating her blood investigations, she was taken for radical hysterectomy with bilateral pelvic lymphadenectomy. Her final histopathology revealed resected tumor free margins, cervical carcinosarcoma with bilateral parametrial tumor deposits. Lymphovascular space invasion was present. Bilateral pelvic nodes were all negative for metastasis. Tumor consisted of sheets and fascicle of pleomorphic plump to spindle cells admixed with multinucleate giant cells and severe atypical cells, with intervening stroma consisting of osteoid like material. On IHC analysis tumor cells were positive for vimentin, p16, SATB2, CK , SMA and negative for CD18, S100, Desmin, p53, CD34. Ki 67 proliferation index was 70%. Her postoperative period was uneventful and she was discharged on the next day with per urethral catheter which was subsequently removed after 21 days. She has been planned for concurrent chemoradiation for her bulky cervical tumor after taking an opinion from a medical oncologist and radiotherapist. Its been 2 months post surgery, she has been compliant in her therapy.
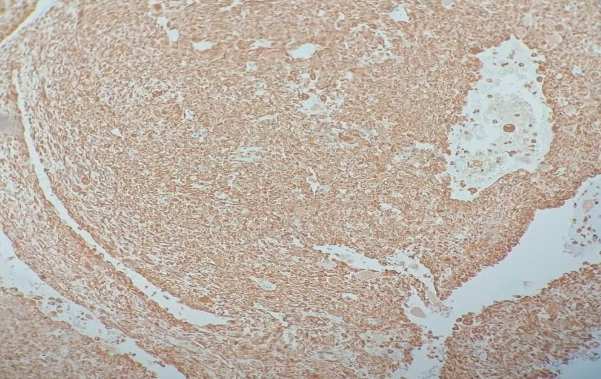
Histopathological figures of our case has been shown.
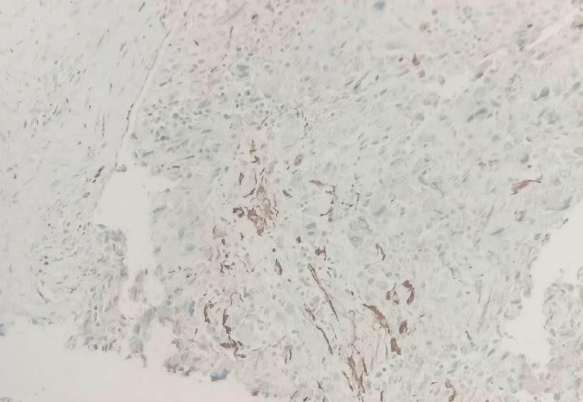
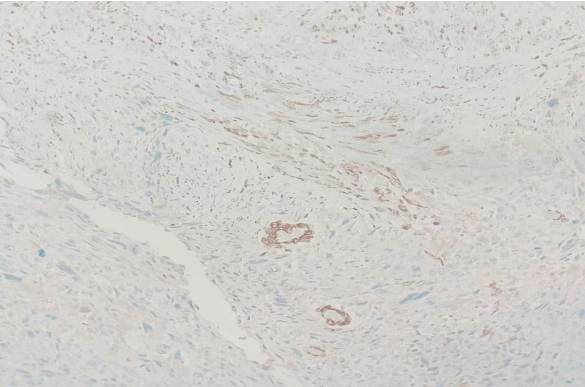
Fig 1
IHC IN CERVICAL BIOPSY SPECIMEN
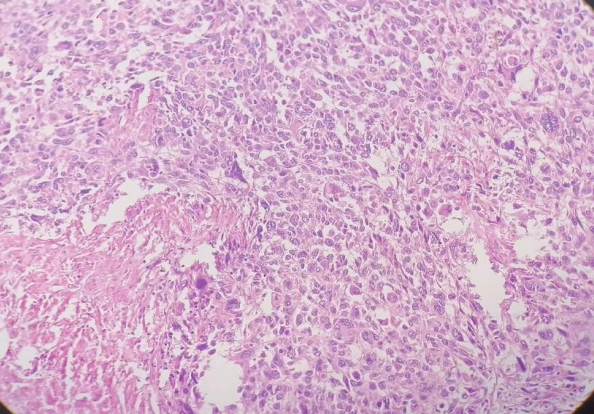
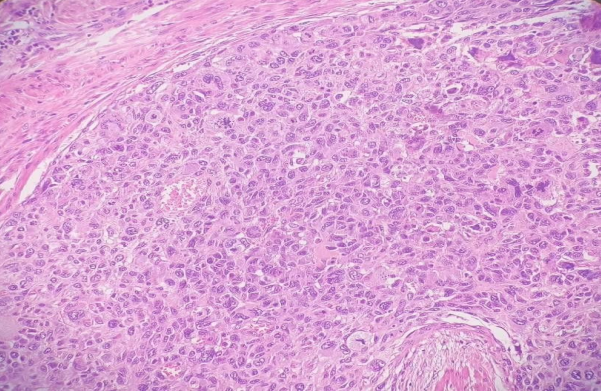
Fig 2, 3, 4
SHOWING EPITHELIAL AND MESENCHYMAL COMPONENTS ON HEMATOXYLIN AND EOSIN STAIN, 400 MAGNIFICATION
Fig 5
Discussion
CCS accounts for nearly half of cervical sarcoma, with the recurrence rate less than 1% of all cervical malignancies (1). The age of initial diagnosis of CCS ranges from 12 to 94 and the vast majority of patients occurred in postmenopausal. Black race, pelvic radiotherapy, chemotherapy, history and HPV infection in particular 16 type are at high risk of developing CCS.
Some studies have documented the integration of high-risk HPV in both epithelial and mesenchymal components of CCS, although the role of HPV in the evolution of CCS needs further exploration. The initial symptoms of CCS are similar to cervical cancer, mainly including abnormal vaginal spotting/bleeding and watery vaginal discharge. Early metastasis in CCS commonly occurs in vagina, rectum, bladder and bones.
CCS usually presents as a polypoid or bulky mass in cervix on ultrasound. CT and MRI can also aid in diagnosis. Histologically, CCS is characterized as a biphasic admixture of intimately juxtaposed carcinomatous and sarcomatous components (2). The malignant epithelial components usually presents with squamous cell carcinoma (SCC), adenocarcinoma of which SCC and adenocarcinoma account for the most. The mesenchymal components, either homologous (gynecologic tissue) or heterologous (represented by osteo sarcomatous components), predominantly presents as long, spindle cell element (3).
Immunohistochemistry (IHC) displays that the epithelial elements mainly express cytokeratin (CK), broad-spectrum cytokeratin marker (MNF-116), low- molecular-weight cytokeratin marker (CAM 5.2), high- molecular-weight cytokeratin marker (34βE12) and epithelial membrane antigen (EMA), whereas the sarcoma components always express vimentin, desmin, smooth muscle-specific actin (SMA) and muscle- specific actin (MSA). Notably, sarcomatous components can express MNF-116, EMA and epithelial components are immunoreactive for vimentin.
Misdiagnosis of CCS sometimes happens in cervical samples because the sarcomatous components are hard to be identified. A meta-analysis showed that the accurate diagnosis rate of CCS by preoperative pathology was only around 56%, and CCS was easy to be misdiagnosed as purely epithelial carcinoma like in our case. (2)
There is no evidence-based guideline on optimal treatment approaches for CCS patients given its rarity. Differential diagnoses of carcinosarcoma of the cervix are differentiated and undifferentiated carcinomas and sarcomatoid carcinoma (1).
Treatment has given priority to surgery. For early-stage CCS patients, surgery combined with adjuvant radiotherapy with or without chemotherapy, is associated with an improved disease- free survival (DFS) and overall survival (OS)(2).
Radiotherapy is commonly recommended to locally advanced-stage CCS patients while chemotherapy is usually conducted in metastatic CCS patients. Radical surgery is the strongest recommendation for FIGO IB stage patients, who account for nearly half of all CCS patients. The common sites for tumor metastasis in cervical carcinosarcoma are the pelvic and paraaortic lymph nodes, peritoneum and the lungs(5). In our case we also preferred surgery as clinical examination was indicating presence of localized lesion with no suspicious nodes.
Conclusion
In conclusion, primary carcinosarcoma of the cervix is a very rare subtype of cervical malignancy. The need for immunohistochemical analysis for its diagnosis can’t be denied. Most of the cases have been treated with primary surgery and we recommend the same as our case. As data on this subtype is rare there is an emerging need for clinical trials so as to decipher diagnosis and management.
References
1.Carcinosarcoma of the Cervix: A Case Report Birendra Bhagat,1 Bijaya Chandra Aacharya,2 Sarita Gurung,2 Ranjan Raj Bhatta,3 Aasiya Rajbhandari4
2.Shu X, Zhou Y, Wei G, Chen X, Qiu M. Cervical Carcinosarcoma: Current Understanding on Pathogenesis, Diagnosis, Management and Future Perspectives. Clinical Medicine Insights: Oncology. 2021;15. doi:10.1177/11795549211056273
3.Lin Y, Chen H, Ye Z, Ding L, Cao Q, Xue L. Synchronous carcinosarcoma of the uterine cervix with adenoid basal carcinoma and cervical intraepithelial neoplasia III: a case report and literature review. Pathol Res Pract. 2017;213:570- 573.
4.Barker HE, Scott CL. Genomics of gynaecological carcinosarcomas and future treatment options. Semin Cancer Biol. 2020;61:110-120.
5.Bansal S, Lewin SN, Burke WM, et al. Sarcoma of the cervix: natural history and outcomes. Gynecol Oncol. 2010;118:134-138.
6. Ribeiro-Silva A, Novello-Vilar A, Cunha-Mercante AM, De Angelo Andrade LA. Malignant mixed mullerian tumor of the uterine cervix with neuroendocrine differentiation. Int J Gynecol Cancer. 2002;12:223-227.