Endoscopic Transpapillary Gallbladder Drainage in a Patient with Recurrent Cholecystitis with Ascites
Endoscopic Transpapillary Gallbladder Drainage in a Patient with Recurrent Cholecystitis with Ascites
Corey Mealer, BS1*, Manjakkollai P. Veerabagu, MD2
1. Medical Student, College of Medicine; Medical University of South Carolina.
2. Affiliate Associate Professor; Medical University of South Carolina.
*Correspondence to: Corey Mealer, BS, Medical Student, College of Medicine; Medical University of South Carolina.
Copyright
© 2025 Corey Mealer, BS, This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 25 July 2025
Published: 04 Aug 2025
Abstract:
Gallbladder disease is typically managed with laparoscopic cholecystectomy in patients who are suitable surgical candidates. For those patients who are not surgical candidates, alternative drainage methods such as percutaneous cholecystostomy (PC) or endoscopic approaches, namely transpapillary gallbladder drainage (ET-GBD) and endoscopic ultrasound guided gallbladder drainage (EUS-GBD), are available. In patients with ascites, ET-GBD is preferred over PC and EUS-GBD due to its lower risk of peritoneal leak and infection. Moreover, ET-GBD is a safe and effective option that preserves normal anatomy and allows for future gallbladder surgery in patients who improve to become surgical candidates. Recent studies support the use of ET-GBD over PC for improved patient comfort, fewer complications, and improved clinical success rates. This case report describes a 67-year-old man with advanced liver cirrhosis, ascites, and recurrent cholecystitis who was successfully treated with ET-GBD.
Endoscopic Transpapillary Gallbladder Drainage in a Patient with Recurrent Cholecystitis with Ascites
Case
The patient is a 67-year-old male with a past medical history of cirrhosis and portal hypertension with ascites due to alcohol and untreated, chronic hepatitis C infection, thrombocytopenia, active tobacco use, coronary artery disease on prasugrel, congestive heart failure, and chronic obstructive pulmonary disease presented with severe right upper quadrant abdominal pain. He was recently hospitalized for a similar complaint, during which workup showed acute cholecystitis. Due to significant comorbidities, active thrombocytopenia, and prasugrel use on the day of admission, he was managed conservatively with antibiotic, amoxicillin-clavulanate. Three weeks later, he returned to the emergency department with the same recurrent pain and tenderness localized to the right upper quadrant, an abdominal fluid wave, and mild jaundice. Repeat CT of the abdomen demonstrated persistently inflamed gallbladder with wall thickening, cystic duct dilatation, abnormal enhancement along the fundus of the gallbladder, and worsening abdominal ascites consistent with acute on chronic cholecystitis. After multi-disciplinary discussion, the patient was deemed a poor surgical candidate. Transpapillary gallbladder drainage (ET-GBD) was deemed the most appropriate non-surgical option given his overall health. Percutaneous gallbladder drainage (PC) and endoscopic ultrasound-guided GBD (EUS-GBD) with a lumen apposing metal stent (LAMS) were not recommended due to the presence of large-volume, recurrent ascites despite ongoing paracentesis and diuretic therapy. Informed consent was obtained for endoscopic retrograde cholangiopancreatography (ERCP) and ET-GBD.
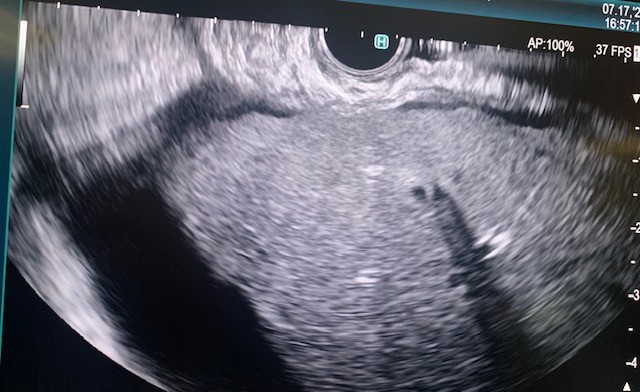
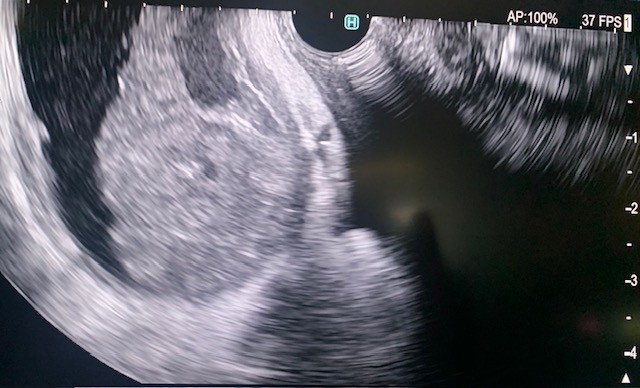
The gastroscope (Olympus, America) was passed into the esophagus under direct visualization. The esophageal and gastroesophageal junction appeared fine without inflammation or lesions. Examination of the stomach showed portal hypertension gastropathy without gastric varices. The duodenal bulb and second part of the duodenum were also clear without pathology. Next, the linear array echoendoscope (Olympus) was passed into the esophagus and advanced into the stomach. EUS imaging revealed a cirrhotic liver surrounded by a large amount of ascites (Fig 1). From the duodenum, the gallbladder was assessed and demonstrated signs of gallbladder wall thickening with a large burden of sludge and small stones (Fig 2).
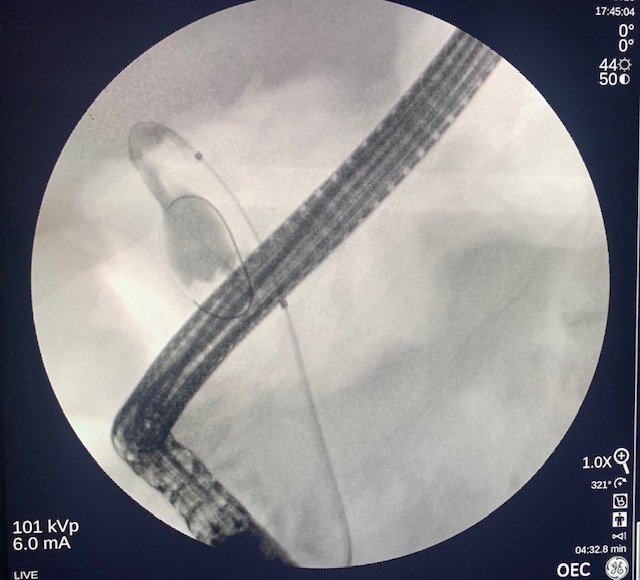
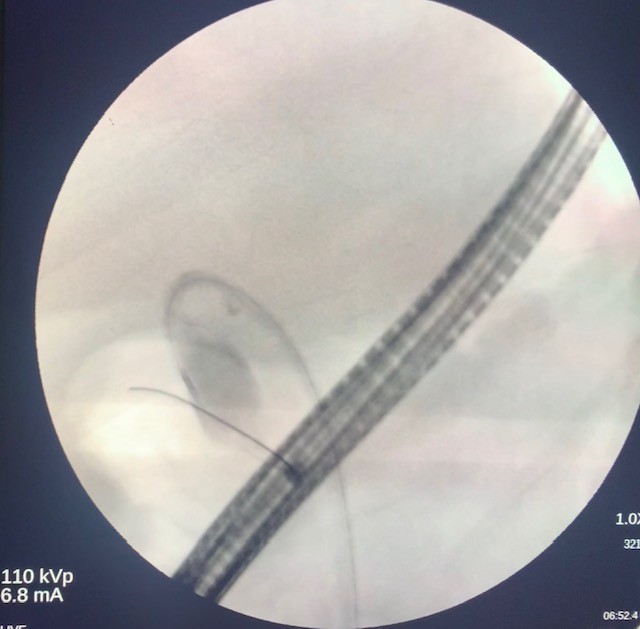
The side-viewing duodenoscope (Olympus) was passed to the duodenum and the major ampulla appeared normal in the second part of the duodenum. A Jagtome Rx 39 (Boston Scientific) with a loaded revolution Jagwire 0.025 was used to cannulate the common bile duct. Cholangiogram showed a very small bile duct, cystic duct and that the gallbladder was filled with a large amount of sludge. Sphincterotomy was performed over the guidewire, and a black pigmented stone was dislodged from the ampulla. Then the Jagwire was maneuvered into the gallbladder the cystic duct was dilated using a Hurricane balloon 4mm x 4cm (Boston Scientific) (Fig 3). A 7 french 15-cm double pig-tail plastic stent (Boston Scientific) was deployed, the proximal curled pigtail in the duodenum and the distal pigtail end was in the gallbladder through the ampulla (Fig 4). The contrast from the gallbladder was subsequently emptied out of the stent showing patency.
The patient reported significant pain relief the next day. He received three paracenteses over his hospitalization obtaining 9,900mL of fluid and a referral for continued outpatient management was placed. He was discharged three days after ET-GBD completion with resolution of his symptoms.
Discussion
Gallbladder diseases have historically been treated with laparoscopic cholecystectomy in surgical candidates and percutaneous cholecystostomy (PC) in non-operable patients. PC is a radiologic procedure where a catheter is placed in the gallbladder under imaging guidance that allows for external drainage of the gallbladder’s contents through the abdominal wall. In addition to drainage, this procedure allows for extraction of stones and the use of stone dissolution therapy1. While the benefits of this procedure include local anesthesia over general anesthesia and cost-effectiveness, the response rate is highly variable, requires maturation before it can be removed, has a high rate of catheter displacement, and results in a poor quality of life for the patient 2.
Fortunately, several endoscopic procedures have become available in recent years to drain the gallbladder and ultimately treat disease including ET-GBD and EUS-GBD 3. These procedures offer less invasive, safe means of draining the gallbladder that can be particularly useful in non-surgical patients with extensive inflammatory changes of the gallbladder. Additionally, these procedures allow for treatment of gallbladder disease in high-risk patients, while preserving quality of life and the comfort of the patient. Contraindications to all endoscopic procedures of the gallbladder include gallbladder perforation or biliary peritonitis 4.
ET-GBD is a safe ERCP procedure where a transpapillary, transcystic stent is placed to create a direct, patent connection between the gallbladder and the duodenum 5. This stent can be used as a bridge to cholecystectomy while the patient is medically optimized or remain as permanent drainage. This procedure has been around since the 1980s, however it was typically avoided due to technical difficulty advancing the stent. However, now cholangioscopy (Spyglass Boston Scientific) can be used in difficult cases to assist in guidewire placement and to confirm the anatomy of the cystic duct and common bile duct. The adverse events of ET-GBD are very similar to anyone undergoing ERCP, such as bleeding, pancreatitis, perforation, and cholestasis, plus adverse events unique to ET-GBD, such as distal biliary tree perforation and stent migration.
In EUS-GBD, an EUS endoscope is advanced into the stomach and duodenum to assess the anatomy and identify an optimal access point. A cautery-enhanced method is preferred using a hot Axios stent Lumen Apposing Metal stent (Boston Scientific). Access from the duodenum is preferred to the stomach to minimize complications. Once the gallbladder is accessed, a guidewire is introduced, followed by the deployment of a lumen apposing metal stent (LAMS)6. The distal flange of the stent is deployed within the gallbladder and pulled snug against its wall, after which the proximal flange is deployed in the duodenum, creating a secure, artificial communication which over time becomes a mature fistula. This fistula provides a wide lumen for effective drainage while minimizing the risk of bile leakage or stent migration due to the anchoring flanges 7. Compared to PC, EUS-GBD offers superior patient comfort and quality of life, while offering a safe and clinically successful procedure for the management of cholecystitis in non-operative patients. While EUS-GBD has been shown to have clinical and technical success benefits over ET-GBD, ET-GBD is preferred over EUS-GBD in patients with coagulopathy, ascites, or who should avoid anatomy altering procedures such as liver transplantation 8.
There is a broad armamentarium for managing gallbladder disease in high-risk surgical candidates with the introduction of these novel endoscopic procedures. ET-GBD is a safe, effective procedure that can serve as a permanent therapy or a bridge therapy for patients who may become surgical candidates since the anatomy has not been altered by this procedure. Additionally, ET-GBD is a superior intervention in patients with concomitant ascites or coagulopathy. More large-scale trials are required to assess the safety and efficacy in different patient populations with these novel endoscopic procedures.
Figure 1. Endoscopic ultrasound of the liver with significant ascites present.
Figure 2. EUS visualization of the gallbladder with stones and a build-up of biliary sludge.
Figure 3. Fluoroscopic evaluation of balloon progression through the common bile duct and cystic duct.
Figure 4. Fluoroscopic imaging of the distal end of the stent deployed in the gallbladder.
References
1. Akhan, O., Akinci, D., Ozmen, M.N. Percutaneous cholecystostomy. Eur J Radiol. 2002;43(3):229-236.
2. Horn, T., Christensen, S.D., Kirkegård, J., Larsen, L.P., Knudsen, A.R., Mortensen, F.V. Percutaneous cholecystostomy is an effective treatment option for acute calculous cholecystitis: a 10-year experience. HPB (Oxford). 2015;17:326-331.
3. Storm, A.C., Vargas, E.J., Chin, J.Y., Chandrasekhara, V., Abu Dayyeh, B.K., Levy, M.J., Martin, J.A., Topazian, M.D., Andrews, J.C., Schiller, H.J., Kamath, P.S., Petersen, B.T. Transpapillary gallbladder stent placement for long-term therapy of acute cholecystitis. Gastrointest Endosc. 2021;94(4):742-748.e1.
4. Saumoy, M., Yang, J., Bhatt, A., Bucobo, J.C., Chandrasekhra, V., Copland, A.P., Krishnan, K., Kumta, N.A., Law, R.J., Pannala, R., Parsi, M.A., Rahimi, E.F., Trikudanathan, G., Trindade, A.J., Lichtenstein, D.R. Endoscopic therapies for gallbladder drainage. Gastrointest Endosc. 2021;94(4):671-684.
5. Lee, T.H., Park, D.H., Lee, S.S., Seo, D.W., Park, S.H., Lee, S.K., Kim, M.H., Kim, S.J. Outcomes of endoscopic transpapillary gallbladder stenting for symptomatic gallbladder diseases: a multicenter prospective follow-up study. Endoscopy. 2011;43(8):702-708.
6. Irani, S.S., Shazrehi, K., Siddiqui, U.D. AGA Clinical Practice Update on Role of EUS-Guided Gallbladder Drainage in Acute Cholecystitis: Commentary. Clinical Gastroenterology and Hepatology. 2023;21(5):1141-1147
7. Itoi, T., Coelho-Prabhu, N., Baron, T. Endoscopic gallbladder drainage for management of acute cholecystitis. Gastrointest Endosc. 2010;71(6):1038-1045.
8. Podboy, A., Yuan, J., Stave, C.D., Chan, S.M., Hwang, J.H., Teoh, A.Y.B. Comparison of EUS-guided endoscopic transpapillary and percutaneous gallbladder drainage for acute cholecystitis: a systematic review with network meta-analysis. Gastrointest Endosc. 2021;93(4):797-804.